![]() ГЄЩГРэЙЄДѓбЇБИПЮжН
ГЄЩГРэЙЄДѓбЇБИПЮжН
![]()
ЮоЛњЛЏбЇBНВИх
32ПЮЪБ
НЬВФЃКЬьНђДѓбЇЕк5Ац
ЫЎРћбЇдК24МЖ1-2Ар
ТоЮЌЮГ
ЛЏбЇЛЏЙЄбЇдК
2024-2025ЃЈ1ЃЉ
ЕквЛеТ ЛЏбЇЗДгІжаЕФжЪСПЙиЯЕКЭФмСПЙиЯЕ
ЪзЯШЃЌЮвУЧЛиЙЫЯТИпжабЇЯАЕФжЊЪЖЃКРэЯыЦјЬхзДЬЌЗНГЬЪНЁЃ
ЦјЬхЕФзюЛљБОЬиеїЃКОпгаПЩбЙЫѕадКЭРЉЩЂадЁЃШЫУЧНЋЗћКЯРэЯыЦјЬхзДЬЌЗНГЬЪНЕФЦјЬхЃЌГЦЮЊРэЯыЦјЬхЁЃРэЯыЦјЬхЗжзгжЎМфУЛгаЯрЛЅЮќв§КЭХХГтЃЌЗжзгБОЩэЕФЬхЛ§ЯрЖдгкЦјЬхЫљеМгаЬхЛ§ЭъШЋПЩвдКіТдЁЃ
РэЯыЦјЬхзДЬЌЗНГЬЪНЃК
pV = nRT R----ФІЖћЦјЬхГЃСП
дкSTPЯТЃЌp =101.325kPa, T=273.15K
n=1.0 molЪБ, Vm=22.414L=22.414ЁС10-3m3
R=8.314 kPa-1mol-1
РэЯыЦјЬхзДЬЌЗНГЬЪНЕФгІгУЃКМЦЫуpЃЌVЃЌTЃЌnЫФИіЮяРэСПжЎвЛЁЃгІгУЗЖЮЇЃКЮТЖШВЛЬЋЕЭЃЌбЙСІВЛЬЋИпЕФецЪЕЦјЬхЁЃ
ЦјЬхЛьКЯЮяжаЦфвЛзщЗжЦјЬхBЖдЦїБкЫљЪЉМгЕФЕЅЮЛУцЛ§ЩЯЕФбЙСІЃЌГЦЮЊИУЦјЬхЗжбЙ(PB)ЃЌЫќЕШгкЯрЭЌЮТЖШЯТЃЌИУЦјЬхЕЅЖРеМгагыЛьКЯЦјЬхЯрЭЌЬхЛ§ЪБЫљВњЩњЕФбЙСІЁЃЛьКЯЦјЬхЕФзмбЙСІЕШгкИїзщГЩЦјЬхЗжбЙСІжЎКЭЃЌДЫОбщЙцдђГЦЕРЖћЖйЗжбЙЖЈТЩЁЃ
ЛЏбЇЗДгІжаЛЙгаЦфЫћКмЖржЪСПЙиЯЕЃЌБШШчХЈЖШЃЌЕШЕШЃЌетРяЮвУЧОЭВЛвЛвЛЛиЙЫСЫЁЃ
ЮЊСЫИќзМШЗЕФЬжТлКЭМЦЫуЛЏбЇЗДгІЕФФмСПБфЛЏЃЌЮвУЧЛЙашвЊСЫНтЛЏбЇЗДгІМЦСПЪНКЭЗДгІНјЖШ
ЛЏбЇЗДгІМЦСПЪНЃКИљОнжЪСПЪиКуЖЈТЩЃЌгУЙцЖЈЕФЛЏбЇЗћКХКЭЛЏбЇЪНРДБэЪОЛЏбЇЗДгІЕФЪНзгЃЌгжНазіЛЏбЇЗДгІЗНГЬЪНЁЃ
ЛЏбЇМЦСПЪ§ЃКЛЏбЇЪНЧАЕФЁАЯЕЪ§ЁБЁЃвдІЭBБэЪОЮяжЪBЕФЛЏбЇМЦСПЪ§ЁЃЙцЖЈЃКЖдгкЗДгІЮяЃЌІЭBЮЊИКЃЌЖдгкВњЮяЃЌІЭBЮЊе§ЁЃІЭBЮЊСПИйвЛЕФСПЁЃ
Р§ШчЃКЗДгІ aA+bBЁњyY+zZ
ПЩаДГЩ -ІЭAA-ІЭBB=ІЭYY+ІЭZZ
МДЃК ІЭA=-aЃЌІЭB=-bЃЌІЭY=yЃЌІЭZ=z
ЩЯЪНПЩМђаДГЩЃК ![]()
ДЫЪНжаЕФBДњБэЗДгІЮяКЭВњЮяЁЃ
ЗДгІНјЖШЃЈІЮЃЉЃКБэЪОЗДгІНјааЕФГЬЖШЛђЮяжЪБфЛЏНјеЙЕФГЬЖШЃЌГЃгУЗћКХІЮБэЪОЃЌЦфЖЈвхЮЊЃК
ІЮ= ![]()
ЪНжаЃЌnB(0)ЮЊЗДгІЦ№ЪМЪБПЬЃЌМДЗДгІНјЖШІЮ=0ЪБЃЌBЕФЮяжЪЕФСПЃЛnBЃЈІЮЃЉЮЊЗДгІНјааЕНtЪБПЬЃЌМДЗДгІНјЖШІЮ=ІЮЪБЃЌBЕФЮяжЪЕФСПЁЃЯдШЛЃЌЗДгІНјЖШІЮЕФЕЅЮЛЮЊmolЁЃ
Р§ШчЃК ЗДгІ N2(g)+3H2(g) ![]() 2NH3(g) ІЮ
2NH3(g) ІЮ
Ц№ЪМЪБnB/mol 3.0 10.0 0 0
t ЪБ nB/mol 2.0 7.0 2.0 ІЮ
ІЮ= ![]() =
= ![]() =
= ![]()
= ![]() =
= ![]() =
= ![]() =1.0mol
=1.0mol
ІЮ=1.0 mol ЪБЃЌБэУїИУЛЏбЇЗДгІМЦСПЪННјааСЫ1.0 mol ЗДгІЃЌ
МД БэЪО1.0 mol N2КЭ1.0 mol ЕФ 3H2 ЗДгІВЂЩњГЩСЫ1.0 mol ЕФ 2NH3ЁЃ
зЂвтЃК
V ЖдгкЭЌвЛЛЏбЇЗДгІЗНГЬЪНЃЌЮоТлгУЗДгІЮяКЭВњЮяжаЕФФФИіЮяжжЕФЮяжЪЕФСПЕФБфЛЏжЕЁїnBРДЧѓЫуЗДгІНјЖШІЮЃЌНсЙћЖМЪЧЯрЭЌЕФЁЃ
V ЗДгІНјЖШІЮЪЧгыЛЏбЇЗДгІМЦСПЪНЯрЦЅХфЕФЁЃ
Р§ШчЃКШєЩЯЗДгІМЦСПЪНИФаДЮЊЃК
![]()
дђtЪБЃЌ ІЮ= ![]() =
= ![]() =2.0mol
=2.0mol
ЛЏбЇЗДгІЕФФмСПЙиЯЕЃКЮвУЧЖМжЊЕРЃЌШЫРрЕФЩњВњЃЌЩњЛюжаЖМРыВЛПЊФмСПЃЌДгЙХДњЕФзъФОШЁЛ№ЃЌЕНКѓУцЕФЛЏЪЏФмдДЃЌМАНёЬьЬсГЋЕФЧхНрФмдДЃЌШЫРрвЛжБдкВЛЖЯЬНЫїКЭЗЂЯжаТЕФФмдДЁЃгааЇРћгУФмСПЃЌПЊЗЂаТЕФФмдДЃЌЪЧЕБНёЩчЛсПЩГжајЗЂеЙЕФБиШЛвЊЧѓЃЌвВЪЧЮвУЧЛЏбЇбЇзгЕФЗмЖЗФПБъжЎвЛЁЃ
ДгЪЎОХЪРМЭПЊЪМЃЌШЫУЧдкбаОПШчКЮЬсИпШШЛњаЇТЪЕФЪЕМљжаЃЌЬсГіСЫШШСІбЇЕФЛљБОРэТлЁЃШчШШСІбЇЕФШ§ДѓЖЈТЩЁЃдкетвЛеТЕФбЇЯАжаЃЌЮвУЧЪзЯШЛсЪьЯЄШШСІбЇЪѕгяМАЯрЙиИХФюЃЌЪьЯЄШШСІбЇЕквЛЖЈТЩЃЌеЦЮеЛЏбЇЗДгІШШаЇгІЃЛеЦЮеИЧЫЙЖЈТЩМАЗДгІШШЕФМЦЫуЁЃ
НгЯТРДЃЌЮвУЧРДбЇЯАШШСІбЇЪѕгяКЭЛљБОИХФюЪзЯШЪЧЃЌЯЕЭГКЭЛЗОГ
ЯЕЭГЃКИљОнШШСІбЇбаОПЕФашвЊДгжмЮЇЕФЮяжЪЪРНчжаШЫЮЊЕиЛЎЗжГіРДЕФвЛВПЗжГЦЮЊШШСІбЇЯЕЭГЃЌМђГЦЯЕЭГЃЌЪЕМЪЩЯОЭЪЧШШСІбЇЕФбаОПЖдЯѓЁЃЯЕЭГЪЧгЩДѓСПЮЂЙлСЃзгЃЈдзгЁЂЗжзгКЭРызгЕШЃЉзщГЩЕФКъЙлМЏКЯЬхЁЃЯЕЭГОпгаБпНчЃЌетвЛБпНчПЩвдЪЧЪЕМЪЕФНчУцвВПЩвдЪЧШЫЮЊШЗЖЈЕФгУРДЛЎЖЈбаОПЖдЯѓЕФПеМфЗЖЮЇЁЃ
ЛЗОГЃКГ§ЯЕЭГвдЭтЃЌгыЯЕЭГгаЯрЛЅзїгУЕФвЛЧаЮяжЪЖМГЦЮЊЛЗОГЃЌВЛЙ§ЃЌЮвУЧЭљЭљжЛНЋгыЯЕЭГУмЧаЯрЙиЕФФЧВПЗжЮяжЪзїЮЊЛЗОГЁЃ
Р§ШчЃКЮвУЧвЊбаОПФГШнЦїжаЫсМюШмвКЕФжаКЭЗДгІЃЌШнЦїжаЕФЫсМюЛьКЯвКГЦЮЊЯЕЭГЃЌЖјШмвКвдЭтЕФВПЗжГЦжЎЮЊЛЗОГЁЃШєШнЦїЪЧГЈПЊЕФЃЌЮвУЧвВПЩвдНЋећИіШнЦїВПЗжзїЮЊЯЕЭГЃЌМДАќКЌЗДгІвККЭвКУцЩЯЕФВПЗжЛьКЯПеЦјЁЃ
ИљОнЯЕЭГКЭЛЗОГжЎМфЮяжЪЁЂФмСПДЋЕнЧщПіЕФВЛЭЌЃЌНЋЯЕЭГЗжЮЊвдЯТШ§жжЧщПіЃК
ЁёГЈПЊЯЕЭГЃКЯЕЭГгыЛЗОГжЎМфЭЈЙ§БпНчМШгаЮяжЪЕФДЋЕнЃЌгжгаФмСПЕФДЋЕнЁЃ
здШЛНчжаЫљгаЕФЩњЮяЬхЯЕЖМПЩПДзїГЈПЊЬхЯЕЃЌвВГЦЮЊПЊЗХЯЕЭГЁЃ
ЁёЗтБеЯЕЭГЃКЯЕЭГгыЛЗОГжЎМфЭЈЙ§БпНчжЛгаФмСПЕФДЋЕнЃЌЖјУЛгаЮяжЪЕФДЋЕнЁЃ
Р§ШчЃКдквЛИіУмБеШнЦїжаНјааН№ЪєФЦгыЫЎзїгУЩњГЩЧтбѕЛЏФЦКЭЧтЦјВЂЗХГіШШСПЕФЗДгІЃЌШєИУШнЦїВЛШУШЮКЮЮяжЪвнЩЂКЭМгШыЃЌЕЋШнЦїВЛЪЧОјШШЕФЃЌГ§здЩэЮТЖШЩ§ИпЭтЃЌЛЙФмНЋШШСПДЋЕнЕНжмЮЇЕФПеЦјжаЃЌдђетИіШнЦїжаЕФЫљгаЮяжЪПЩвдЭГПДЮЊвЛИіЗтБеЬхЯЕЁЃ
ЁёИєРыЯЕЭГЃКЯЕЭГгыЛЗОГжЎМфУЛгаШЮКЮзїгУЁЃМШУЛгаЮяжЪЕФДЋЕнЃЌвВУЛгаФмСПЕФНЛЛЛЁЃ
ЪЕМЪЩЯЃЌОјЖдИєРыЕФЯЕЭГЪЧВЛДцдкЕФЃЌЕЋгЩгкбаОПЕФашвЊЃЌЮвУЧГЃГЃНЋвЛаЉЯЕЭГНќЫЦЕиПДзїИєРыЯЕЭГЁЃШчЧАЪіЕФУмБеШнЦїЛЛГЩОјШШЕФВФСЯЃЌМйЩшЛЙВЛЗјЩфКЭЮќЪеФмСПЃЌдђетбљЕФЯЕЭГОЭПЩПДзїЪЧИєРыЯЕЭГЁЃДЫЭтЃЌЯЕЭГгыЛЗОГМгЦ№РДЃЌвВПЩвдПДзївЛИіДѓЕФИєРыЯЕЭГЁЃ
зДЬЌКЭзДЬЌКЏЪ§
ЬхЯЕЕФзДЬЌЃКЪЧЬхЯЕЮяРэаджЪКЭЛЏбЇаджЪЕФзлКЯБэЯжЁЃетаЉаджЪЖМЪЧКъЙлЕФЮяРэСПЃЌЙЪгжГЦЮЊЬхЯЕЕФКъЙладжЪЃЌШчЮТЖШЃЈTЃЉЁЂбЙЧПЃЈpЃЉЁЂЬхЛ§ЃЈVЃЉЁЂЮяжЪЕФСПЃЈnЃЉЁЂжЪСПЃЈmЃЉЁЂУмЖШЃЈІбЃЉЕШЕШЁЃЕБЬхЯЕЕФКъЙладжЪЖМОпгаШЗЖЈЕФЪ§жЕЖјЧвВЛЫцЪБМфЖјБфЛЏЪБЃЌЬхЯЕОЭДІдквЛЖЈЕФШШСІбЇзДЬЌЃЌМђГЦзДЬЌЁЃ
зДЬЌКЏЪ§ЃКШєЬхЯЕЕФКъЙладжЪБфСЫЃЌзДЬЌвВОЭЫцжЎЖјБфЃЌБфЛЏЧАЕФзДЬЌГЦЮЊЪМЬЌЃЈinitial stateЃЉЃЌБфЛЏКѓЕФзДЬЌГЦЮЊжеЬЌ(final state)ЁЃЬхЯЕЕФетаЉКъЙладжЪгыЬхЯЕЕФзДЬЌжЎМфДцдкЖдгІЕФКЏЪ§ЙиЯЕЁЃУшЪіЬхЯЕзДЬЌЕФетаЉКъЙладжЪгжБЛГЦЮЊзДЬЌКЏЪ§ЁЃЧАУцЬсЕНЕФЮяРэСПTЁЂpЁЂVЁЂnЁЂmЁЂІбЕШЖМЪЧзДЬЌКЏЪ§ЁЃБОеТЛЙНЋНщЩмвЛаЉаТЕФзДЬЌКЏЪ§ЁЃ
зДЬЌКЏЪ§ЕФжївЊЬиЕуЃК
ЂХзДЬЌвЛЖЈЃЌзДЬЌКЏЪ§вЛЖЈЃЛ
ЂЦзДЬЌБфЛЏЃЌзДЬЌКЏЪ§вВЫцжЎБфЛЏЃЌЧвзДЬЌКЏЪ§ЕФБфЛЏжЕжЛгыЪМЬЌЁЂжеЬЌгаЙиЃЌЖјгыБфЛЏЭООЖЮоЙиЁЃ
ЂЧЯЕЭГЕФаджЪжЎМфгавЛЖЈЕФСЊЯЕЃЌР§ШчЃКpV = nRT,ОЭУшЪіСЫРэЯыЦјЬхаджЪp,V,T КЭ n жЎМфЕФЙиЯЕЁЃЫљвдУшЪіЯЕЭГзДЬЌЪБЃЌбЁдёБивЊЕФФмШЗЖЈЯЕЭГзДЬЌЕФМИИіаджЪОЭПЩвдСЫЁЃ
зДЬЌКЏЪ§ЕФетМИИіаджЪЃЌдквдКѓЕФЮоЛњЛЏбЇШШСІбЇМЦЫужаЦ№ЕНСЫЗЧГЃживЊЕФзїгУЁЃ
Й§ГЬ
Й§ГЬЃКШШСІбЇЬхЯЕжаЗЂЩњЕФвЛЧаБфЛЏЖМГЦЮЊШШСІбЇЙ§ГЬЃЌМђГЦЙ§ГЬЁЃШчЦјЬхЕФбЙЫѕгыХђеЭЁЂвКЬхЕФеєЗЂгыФ§ЙЬвдМАЛЏбЇЗДгІЕШЕШЖМЪЧШШСІбЇЙ§ГЬЃЌвђЮЊЫќУЧЖМЪЙЬхЯЕЕФзДЬЌЗЂЩњСЫБфЛЏЁЃЕБЯЕЭГДгЪМЬЌЕНжеЬЌЪБЃЌФГаЉЮяжЪЫцЪБМфЗЂЩњвЛЯЕСаИФБфГЦЮЊЙ§ГЬЁЃ
ЭООЖЃКЯЕЭГФкЪМЬЌЕНжеЬЌПЩОгЩВЛЭЌЕФЙ§ГЬЃЌетаЉЙ§ГЬЕФзмКЭГЦЮЊЭООЖЁЃ
ИљОнЙ§ГЬжаЯЕЭГЕФpЃЌVЃЌTБфЛЏЬиЕуЃЌНЋЙ§ГЬЗжЮЊвдЯТШ§жжЧщПіЃК
ЁёЖЈЮТЙ§ГЬЃКЪМЬЌЁЂжеЬЌЕФЮТЖШЯрЕШЃЌВЂЧвЙ§ГЬжаЪМжеБЃГжетИіЮТЖШЁЃ
ЁёЖЈбЙЙ§ГЬЃКЪМЬЌЁЂжеЬЌЕФбЙСІЯрЕШЃЌВЂЧвЙ§ГЬжаЪМжеБЃГжетИібЙСІЁЃ
ЁёЖЈШнЙ§ГЬЃКЯЕЭГЕФЪМЬЌгыжеЬЌШнЛ§ЯрЭЌЃЌВЂЧвЙ§ГЬжаЪМжеБЃГжЭЌбљЕФШнЛ§ЁЃ
Яр
ЯрЃКЯЕЭГжаЮяРэаджЪКЭЛЏбЇаджЪЭъШЋЯрЭЌЕФЧвгыЦфЫћВПЗжгаУїШЗНчУцЗжИєПЊРДЕФШЮКЮОљдШВПЗжЁЃ
ЗжРрЃКОљЯрЯЕЭГ(ЛђЕЅЯрЯЕЭГ)ЁЂЗЧОљЯрЯЕЭГ(ЛђЖрЯрЯЕЭГ)
ШШКЭЙІЪЧЯЕЭГЗЂЩњБфЛЏЪБгыЛЗОГНјааФмСПНЛЛЛЕФСНжжаЮЪНЁЃЯТУцЮвУЧОЭЗжБ№НщЩмвЛЯТЃК
ШШЃКвђЮТЖШВЛЭЌЖјдкЯЕЭГКЭЛЗОГжЎМфДЋЕнЕФФмСПаЮЪНЃЌГЃгУЗћКХQБэЪОЃЌЦфБОжЪЪЧЮяжЪСЃзгЛьТвдЫЖЏЕФКъЙлБэЯжЁЃ
зЂвтЃК
ШШСІбЇжаЙцЖЈЃКЯЕЭГЮќШШQ > 0ЃЛЯЕЭГЗХШШQ < 0ЁЃ
q ВЛЪЧзДЬЌКЏЪ§ЃЌЦфжЕгыОпЬхЕФБфЛЏЭООЖгаЙиЁЃ
ЙІЃКЯЕЭГгыЛЗОГжЎМфвдГ§ШШвдЭтЕФЦфЫћаЮЪНЖјДЋЕнЕФФмСПЃЌГЃгУЗћКХWБэЪОЃЌШчЬхЛ§ЙІЁЂЛњаЕЙІЁЂЕчЙІЁЂБэУцЙІЕШЃЌЦфБОжЪЪЧЮяжЪСЃзгзїЖЈЯђдЫЖЏЕФНсЙћЁЃ
зЂвтЃК
�������� ЛЗОГЖдЯЕЭГзіЙІW > 0ЃЛЯЕЭГЖдЛЗОГзіЙІW < 0ЁЃ
�������� WВЛЪЧзДЬЌКЏЪ§ЃЌЦфжЕгыОпЬхЕФБфЛЏЭООЖгаЙиЁЃ
ЙІПЩвдЗжЮЊЬхЛ§ЙІКЭЗЧЬхЛ§ЙІЃК
ЁёЬхЛ§ЙІЃКгЩгкЯЕЭГЬхЛ§БфЛЏЖјгыЛЗОГНЛЛЛЕФЙІЁЃ
Р§ШчЃКЦјИзжаЦјЬхЕФХђеЭЛђБЛбЙЫѕЁЃШєКіТдСЫЛюШћЕФжЪСПМАЛюШћгыЦјИзБкМфЕФФІВССІЃЌЛюШћНиУцУцЛ§ЮЊA,дкКуЖЈЭтбЙЙ§ГЬжа,ЯЕЭГПЫЗўЭтбЙpex ХђеЭ,ЛюШћвЦЖЏОрРыlЃЌдкЖЈЮТЯТЯЕЭГЖдЛЗОГзіЙІЃЌ
W = - Fexl ЖјFexl = pexЁЄA
W = -pexЁЄAЁЄl= -pexЁЄІЄV = -pex(V1-V2)
ЪНжаV1ЃЌV2ЗжБ№ЮЊХђеЭКѓКЭХђеЭЧАЕФЦјИзЕФШнЛ§ЃЌМДЦјЬхЕФЬхЛ§ЁЃ ПЩМћЃЌдкЖЈШнЙ§ГЬжаЯЕЭГгыЛЗОГжЎМфУЛгаЬхЛ§ЙІЕФНЛЛЛЁЃ
ЁёЗЧЬхЛ§ЙІЃКЬхЛ§ЙІвдЭтЕФЫљгаЦфЫћаЮЪНЕФЙІЮЊЗЧЬхЛ§ЙІЃЌ ШчЕчЙІЁЂБэУцЙІЕШЁЃдкНгЯТРДЕФбЇЯАжаЃЌЮвУЧМйЩшЯЕЭГЕФЗЧЬхЛ§ЙІЮЊСуЁЃ
ШШСІбЇФм
ФмСПЪЧЮяжЪдЫЖЏЕФЛљБОаЮЪНЁЃвЛАуЯЕЭГЕФФмСПЃЌАќРЈвдЯТШ§ИіВПЗжЃК
ЃЈ1ЃЉЖЏФмЁЊгЩЯЕЭГЕФдЫЖЏЫљОіЖЈЕФФмСПЁЃ
ЃЈ2ЃЉЪЦФмЁЊгЩЯЕЭГдкФГвЛЭтСІГЁжаЕФЮЛжУЫљОіЖЈЕФФмСПЁЃ
ЃЈ3ЃЉФкФмЁЊЯЕЭГФкВПЫљгаЮЂЙлСЃзгЫљДЂВиЕФШЋВПФмСПжЎКЭЁЃ
ЮЊСЫМђЛЏЮЪЬтЃЌЛЏбЇШШСІбЇЭЈГЃжЛбаОПОВжЙЕФЬхЯЕЃЌЧвВЛПМТЧЭтСІГЁЕФзїгУЃЌдђШШСІбЇЬхЯЕЕФФмСПЃЌвВОЭНіжИФкФмЖјбдЁЃвђДЫЃЌФкФмгжГЦШШСІбЇФмЁЃ
ШШСІбЇФмЃКдкВЛПМТЧЯЕЭГЕФећЬхЖЏФмКЭЪЦФмЕФЧщПіЯТЃЌЯЕЭГФкВПЫљгаЮЂЙлСЃзгЫљДЂВиЕФШЋВПФмСПжЎКЭЁЃМЧзїUЁЃ
ШШСІбЇФмРДздгкЮЂЙлСЃзгЕФдЫЖЏгыЯрЛЅзїгУЁЃетаЉдЫЖЏгызїгУАќРЈЗжзгЕФЦНЖЏЁЂзЊЖЏКЭеёЖЏЃЌЗжзгМфЕФЯрЛЅЮќв§гыХХГтЃЌЗжзгФкдзгМфЕФЯрЛЅзїгУЃЌдзгФкКЫгыЕчзгЕФЯрЛЅзїгУЃЌКЫФкЛљБОСЃзгЕФЯрЛЅзїгУЕШЁЃЕБЯЕЭГФкВПзщГЩЕФЮяжжЕФЮяжЪЕФСПвдМАФГаЉЬѕМўШЗЖЈжЎКѓЃЌМДЯЕЭГЕФзДЬЌвЛЖЈЪБЃЌЯЕЭГЕФШШСІбЇФмОЭгаШЗЖЈЕФжЕЁЃ
зЂвтЃКШШСІбЇФмЕФОјЖджЕВЛПЩВтЖЈЁЃ
ШШСІбЇФмЃЈUЃЉЪЧзДЬЌКЏЪ§ЃЌЦфБфЛЏСПІЄUгыЭООЖЮоЙиЁЃ
ШШСІбЇЕквЛЖЈТЩ
ШШСІбЇЕквЛЖЈТЩЕФЪЕжЪЪЧФмСПЪиКугызЊЛЏЖЈТЩЁЃМДЗтБеЯЕЭГЗЂЩњзДЬЌБфЛЏЪБЃЌЦфШШСІбЇФмЕФБфЛЏЕШгкЙ§ГЬжаЛЗОГгыЯЕЭГДЋЕнЕФШШгыЙІЕФзмКЭЁЃ
ЗтБеЯЕЭГЕФШШСІбЇЕквЛЖЈТЩЕФЪ§бЇБэДяЪНЃКвЛЗтБеЯЕЭГЃЌДгЪМЬЌЃЈШШСІбЇФмЮЊU1ЃЉБфЛЏЕНЪМЬЌЃЈШШСІбЇФмЮЊU2ЃЉЃЌдкДЫЙ§ГЬжаЃЌЯЕЭГДгЛЗОГЮќШШЮЊQЃЌЭЌЪБЛЗОГЖдЯЕЭГзіЙІЮЊWЃЌДЫЪБЃЌU2 - U1= q + WЃЌМД ІЄU = q + W
ШШСІбЇЕквЛЖЈТЩГЪЯжЦфЬиЪтЕФаЮЪНЃК
ЁёИєРыЯЕЭГЕФЙ§ГЬЃК Q=0ЃЌW=0ЃЌЫљвдІЄU=0ЁЃМДИєРыЯЕЭГЕФШШСІбЇФмЪЧЪиКуЕФЁЃ
ЁёбЛЗЙ§ГЬЃКЯЕЭГгЩЪМЬЌОвЛЯЕСаБфЛЏгжЛиИДЕНдРДзДЬЌЁЃІЄU=0ЃЌQ= -W
зЂвтЃКдкИУПЮГЬжаНёКѓЮвУЧжЛбаОПЗтБеЯЕЭГЁЃ
ьЪБфКЭШШЛЏбЇЗНГЬЪН
1ЁЂьЪКЭьЪБф
здШЛНчжаЃЌвЛАуЕФЙ§ГЬЃЌАќРЈвЛАуЕФЛЏбЇЗДгІЃЌЭљЭљжЛЩцМАЬхЛ§ЙІЃЌКмЩйзіЗЧЬхЛ§ЙІЁЃНіПМТЧЬхЛ§ЙІЪБЃЌШШСІбЇЕквЛЖЈТЩбнБфЮЊ ЁїU = q + W = q Ѓ pЭтЁїV
зЂвтЃКдкИУПЮГЬжаНёКѓЮвУЧНіПМТЧЗтБеЯЕЭГжЛзїЬхЛ§ЙІЕФЧщПіЁЃ
ЗДгІШШЃКдквЛЖЈЬѕМўЯТЃЌЛЏбЇЗДгІЙ§ГЬжаЮќЪеЛђЗХГіЕФШШСПЁЃ
ИљОнЗДгІЬѕМўЕФВЛЭЌЃЌЗДгІШШЗжЮЊЃК
ЁёЖЈШнЗДгІШШ
ЖдгкЗтБеЯЕЭГЃЌдкЖЈШнЙ§ГЬжаЃЌІЄV =0ЃЌЯЕЭГЕФЬхЛ§ЙІW =0ЃЌШєЗЧЬхЛ§ЙІЮЊСуЃЌдђЃКQv = ІЄU
QvЮЊЖЈШнЗДгІШШЁЃМДдкЖЈШнЧвЗЧЬхЛ§ЙІЮЊСуЕФЙ§ГЬжаЗтБеЯЕЭГДгЛЗОГЮќЪеЕФШШЕШгкЯЕЭГЕФШШСІбЇФмЕФдіМгЁЃ
ЁёЖЈбЙЗДгІШШ
дкЖЈбЙЙ§ГЬжаЃЌЬхЛ§ЙІW= -pexІЄVЁЃШєЗЧЬхЛ§ЙІЮЊСуЃЌдђЃЌ ІЄU = Qp - pexІЄV
U2 - U1 =Qp - pex(V2-V1)
вђЮЊ pex = p1 = p2
U2 - U1 = Qp - (p2V2-p1V1)
Qp = (U2 + p2V2)-(U1 + p1V1) = ІЄ(U - pV)
ЖЈвхЃК H = U + pV
ЁїH = H2ЃH1= Qp
вђЪНжаU ЁЂpЁЂV ОљЮЊзДЬЌКЏЪ§ЃЌЙЪHвВЮЊзДЬЌКЏЪ§ЃЌГЦЮЊьЪЃЌЁїHГЦЮЊьЪБфЁЃ
QpЮЊЖЈбЙЗДгІШШЁЃМДдкЖЈбЙКЭВЛзіЗЧЬхЛ§ЙІЕФЙ§ГЬжаЃЌЗтБеЯЕЭГДгЛЗОГЫљЮќЪеЕФШШЕШгкЯЕЭГьЪЕФдіМгЁЃ
зЂвтЃК
�������� ьЪЪЧзДЬЌКЏЪ§ЁЃвђЮЊU ЁЂpЁЂV ОљЮЊзДЬЌКЏЪ§ЃЌЙЪHвВЮЊзДЬЌКЏЪ§ЁЃ
�������� ьЪЪЧвЛИіОпгаФмСПСПИйЕФГщЯѓЕФШШСІбЇКЏЪ§ЃЌЦфБОЩэЕФЮяРэвтвхВЂВЛЯѓШШСІбЇФмUФЧбљУїШЗЁЃ
�������� HЕФОјЖджЕЮоЗЈШЗЖЈЁЃгЩгкЬхЯЕФкФмUЕФОјЖджЕВЛФмШЗЖЈЃЌHЕФОјЖджЕвВЮоЗЈШЗЖЈЁЃ
�������� ШШСІбЇжаЙцЖЈЃКЮќШШЗДгІЁїH > 0ЃЛЗХШШЗДгІЁїH < 0ЁЃ
�������� РэЯыЦјЬхЕФьЪжЛЪЧЮТЖШЕФКЏЪ§ЁЃ
2.ШШЛЏбЇЗНГЬЪН
(1) ЗДгІЕФФІЖћШШСІбЇФмБфІЄrUmКЭЗДгІЕФФІЖћьЪБфІЄrHm
ЛЏбЇЗДгІЕФЗДгІНјЖШБфЛЏСПЮЊІЄІЮЃЈІЄІЮ=ІЮ-0=ІЮЃЉЃЌЗДгІЕФШШСІбЇФмБфКЭьЪБфЗжБ№ЮЊІЄUКЭІЄHЃЌдђЃК
![]()
![]()
втвхЃКЗДгІНјЖШЮЊ1molЪБЃЌШШСІбЇФмЕФБфЛЏСПКЭьЪЕФБфЛЏСПЁЃ
ЂЦБъзМзДЬЌ
ФГаЉШШСІбЇСПЃЈШчШШСІбЇФмUЁЂьЪHЕШЃЉЕФОјЖджЕЪЧЮоЗЈВтСПЕФЃЌЕЋЮвУЧФмВтСПЦфзДЬЌБфЛЏЫљв§Ц№ЕФБфЛЏжЕЃЌвђДЫЃЌЮвУЧОЭвЊЮЊЮяжЪЕФзДЬЌЖЈвхвЛИіЛљЯпЁЃЛђБъзМЬЌОЭЪЧетбљвЛИіЛљЯпЁЃ
БъзМзДЬЌЃКБъзМбЙСІ ![]() =100kPaЯТЮяжЪШЗЧаЕФОлМЏзДЬЌ
=100kPaЯТЮяжЪШЗЧаЕФОлМЏзДЬЌ
ЁёЦјЬЌЕФБъзМЬЌЃКЮТЖШЮЊTЃЌбЙСІЮЊ ![]() ЯТВЂБэЯжГіРэЯыЦјЬхЬиадЕФЦјЬхДПЮяжЪBЃЈМйЯыЃЉзДЬЌЁЃ
ЯТВЂБэЯжГіРэЯыЦјЬхЬиадЕФЦјЬхДПЮяжЪBЃЈМйЯыЃЉзДЬЌЁЃ
ЁёвКЬЌЃЈЛђЙЬЬЌЃЉЕФБъзМЬЌЃКЮТЖШЮЊTЃЌбЙСІЮЊ ![]() ЯТвКЬхЃЈЛђЙЬЬхЃЉДПЮяжЪBЕФзДЬЌЁЃ
ЯТвКЬхЃЈЛђЙЬЬхЃЉДПЮяжЪBЕФзДЬЌЁЃ
ЁёвКЬхШмвКжаШмМСКЭШмжЪЕФБъзМЬЌЃКЮТЖШЮЊTЁЂ бЙСІЮЊ ![]() ЯТ,ШмвКжаШмМСвдДПЮяжЪЕФзДЬЌЮЊБъзМЬЌЃЛШмвКжаЕФШмжЪвджЪСПФІЖћХЈЖШbB=
ЯТ,ШмвКжаШмМСвдДПЮяжЪЕФзДЬЌЮЊБъзМЬЌЃЛШмвКжаЕФШмжЪвджЪСПФІЖћХЈЖШbB= ![]() =1molЁЄkg-1ЃЌВЂБэЯжГіЮоЯоЯЁЪЭШмвКЬиадЪБШмжЪBЕФЃЈМйЯыЃЉзДЬЌЁЃЃЈЮоЛњЛЏбЇжазївдЯТНќЫЦДІРэ
=1molЁЄkg-1ЃЌВЂБэЯжГіЮоЯоЯЁЪЭШмвКЬиадЪБШмжЪBЕФЃЈМйЯыЃЉзДЬЌЁЃЃЈЮоЛњЛЏбЇжазївдЯТНќЫЦДІРэ ![]() Ёж
Ёж ![]() ЃЌbЁжcЃЉ
ЃЌbЁжcЃЉ
зЂвтЃК
БъзМзДЬЌЖдШШСІбЇЮТЖШTЮДзїОпЬхЙцЖЈЁЃВЛЙ§,аэЖрШШСІбЇЪ§ОнЖрЪЧдкT=298.15KЯТЕУЕНЕФЃЌЫљвдШєЮДМгЬиЪтЬсЪО,БОПЮГЬжаЩцМАЕНЕФШШСІбЇКЏЪ§Ољвд298.15KЮЊВЮПМЮТЖШЁЃ
ЂЧШШЛЏбЇЗНГЬ
ШШЛЏбЇЗНГЬЪНЃКБэЪОЛЏбЇЗДгІМАЦфЗДгІШШЃЈБъзМФІЖћьЪБфЃЉЙиЯЕЕФЛЏбЇЗДгІЗНГЬЪН
Р§ШчЃК2H2(g) + O2(g)Ёњ2H2O(g) ![]()
КЌвхЃК
дкЮТЖШЮЊ298.15KЕФЖЈбЙЙ§ГЬжаЃЌжюЦјЬхбЙСІОљЮЊБъзМбЙСІ ![]() (100kPa)ЯТЃЌЗДгІНјЖШЮЊ1molЪБЃЌИУЗДгІЕФБъзМФІЖћьЪБф
(100kPa)ЯТЃЌЗДгІНјЖШЮЊ1molЪБЃЌИУЗДгІЕФБъзМФІЖћьЪБф ![]() ЁЃ
ЁЃ
зЂвтЃК
ЁёзЂУїЮяжЪЕФОлМЏзДЬЌЃЌЛЏбЇЗДгІМЦСПЪНжаИїЮяжЪЕФОлМЏЬЌВЛЭЌЃЌ ![]() ВЛЭЌЁЃ
ВЛЭЌЁЃ
2H2(g) + O2(g)Ёњ2H2O(l) ![]()
ЁёЗДгІМЦСПЪНаыХфЦНЃЌЧв ![]() БиаыгыЗДгІМЦСПЪНЯрЦЅХфЁЃ
БиаыгыЗДгІМЦСПЪНЯрЦЅХфЁЃ
![]()
![]()
ЁёзЂУїЗДгІЮТЖШЃЌЕЋьЪБфЫцЮТЖШИФБфБфЛЏВЛДѓЁЃБОПЮГЬжаЃЌгаЪБЖдГЃЮТГЃбЙЯТЕФШШЛЏбЇЗНГЬЪНВЛзЂУїЗДгІЬѕМўЃЌЖрвд ![]() (298K)БэЪОжЎЁЃ
(298K)БэЪОжЎЁЃ
CH4(g) + H2O ЁњCOЃЈgЃЉ+ 3H2ЃЈgЃЉ ![]()
![]()
3. ІЄrUmгыІЄrHmЕФЙиЯЕ
![]() ИљОнШШСІбЇЕквЛЖЈТЩЃК
ИљОнШШСІбЇЕквЛЖЈТЩЃК
дђ ![]()
ЖдгкЮоЦјЬхВЮМгЕФЗДгІЃКW = ЈCpex ІЄV = 0ЃЌ![]()
ЖдгкгаЦјЬхВЮМгЕФЗДгІЃК
| |
|
|
![]() ЕФЪ§жЕгы
ЕФЪ§жЕгы ![]() ЕФЪ§жЕЯрВюСНИіЪ§СПМЖЃЌЫљвд
ЕФЪ§жЕЯрВюСНИіЪ§СПМЖЃЌЫљвд ![]() гы
гы ![]() ЯрВюЩѕаЁЁЃ
ЯрВюЩѕаЁЁЃ
НсТлЃК ЖдгкЛЏбЇЗДгІ ![]()
БъзМФІЖћЩњГЩьЪЃЈжиЕуЃЉ
БъзМЩњГЩьЪЃКдкЮТЖШTЯТЃЌгЩВЮПМЬЌЕФЕЅжЪЩњГЩЮяжЪB(ІЭB= +1)ЕФЗДгІЕФБъзМФІЖћьЪБфЁЃМЧзїЃК ![]() (B,ЯрЬЌЃЌT), ЕЅЮЛkJЁЄmol-1ЁЃ
(B,ЯрЬЌЃЌT), ЕЅЮЛkJЁЄmol-1ЁЃ
зЂвтЃКБъзМФІЖћЩњГЩьЪЪЧЮяжЪBЕФЩњГЩЗДгІЕФБъзМФІЖћьЪБфЁЃ
ВЮПМзДЬЌЕЅжЪЃКвЛАуЪЧЮТЖШЮЊTЃЌбЙСІЮЊ ![]() ЪБЮяжЪзюЮШЖЈЕФзДЬЌЃЌЕЋИіБ№ЧщПіЯТЃЌАДЯАЙпжИЖЈВЮПМЬЌЁЃ
ЪБЮяжЪзюЮШЖЈЕФзДЬЌЃЌЕЋИіБ№ЧщПіЯТЃЌАДЯАЙпжИЖЈВЮПМЬЌЁЃ
Р§ШчЃК H2(g)ЃЌO2(g)ЃЌЪЏФЋЃЌАзСзP4(s,Аз)ЃЈЖјЪЕМЪЩЯАзСзВЛМАКьСзКЭКкСзЮШЖЈЃЉЁЃ
дкШЮКЮЮТЖШЯТ, ![]() (ВЮПМЬЌЕЅжЪ,T)=0
(ВЮПМЬЌЕЅжЪ,T)=0
ИїжжЮяжЪЕФ ![]() ЖрЪ§аЁгкСу
ЖрЪ§аЁгкСу
ЭЌРраЭЛЏКЯЮяЕФ ![]() гыЮШЖЈадЕФЙиЯЕЃКРћгУЛЏКЯЮяЕФБъзМФІЖћЩњГЩьЪПЩвдБШНЯЯрЭЌРраЭЛЏКЯЮяЕФЯрЖдЮШЖЈадЁЃЛЏКЯЮяЕФБъзМФІЖћЩњГЩьЪдНаЁЃЌЛЏКЯЮядНЮШЖЈЁЃ
гыЮШЖЈадЕФЙиЯЕЃКРћгУЛЏКЯЮяЕФБъзМФІЖћЩњГЩьЪПЩвдБШНЯЯрЭЌРраЭЛЏКЯЮяЕФЯрЖдЮШЖЈадЁЃЛЏКЯЮяЕФБъзМФІЖћЩњГЩьЪдНаЁЃЌЛЏКЯЮядНЮШЖЈЁЃ
HessЖЈТЩ
HessЃЈИЧЫЙЃЉдкДѓСПЪЕбщЕФЛљДЁЩЯзмНсГіЃКЁАвЛИіЛЏбЇЗДгІВЛЙмЪЧвЛВНЭъГЩЛђЪЧЗжМИВНЭъГЩЃЌЫќЕФЗДгІШШЖМЪЧЯрЭЌЕФЁБЁЃ
гЩгкЛЏбЇЗДгІвЛАуЖМдкКубЙЛђКуШнЬѕМўЯТНјааЃЌЖјКубЙЗДгІШШQp=ЁїHЃЌКуШнЗДгІШШQv=ЁїUЃЌHКЭUЖМЪЧзДЬЌКЏЪ§ЃЌЦфЁїHКЭЁїUжЛШЁОігкЪМЬЌКЭжеЬЌЃЌгыЫљОРњЕФЭООЖЮоЙиЁЃ
HessЖЈТЩИќзМШЗЕФБэЪіЮЊЃКдкВЛзїЗЧЬхЛ§ЙІЁЂКубЙЛђКуШнЬѕМўЯТЃЌШЮКЮвЛИіЛЏбЇЗДгІВЛЙмЪЧвЛВНЭъГЩЛЙЪЧЗжМИВНЭъГЩЃЌЦфЗДгІШШжЛОіЖЈгкЬхЯЕЕФЪМЬЌКЭжеЬЌЃЌгыЗДгІЫљОРњЕФЭООЖЮоЙиЁЃ
ИЧЫЙЖЈТЩЪЧШШЛЏбЇМЦЫуЕФЛљДЁЁЃ
HessЖЈТЩЕФгІгУ1ЃК.РћгУШШЛЏбЇЗНГЬЕФзщКЯМЦЫу ![]()
Р§ШчЃКвбжЊ298.15KЯТЃЌЗДгІЃК
Ђй C(s) + O2(g) ЁњCO2(g) ![]() (1)= -393.57kJЁЄmol-1
(1)= -393.57kJЁЄmol-1
Ђк CO(g) + O2(g) Ёњ CO2(g) ![]() (2)=-282.98kJЁЄmol-1
(2)=-282.98kJЁЄmol-1
ЂлМЦЫуЗДгІ ![]()
НтЃКЗДгІЂйПЩвдЭЈЙ§СНжжЭООЖРДЪЕЯж
ИљОнHessЖЈТЩЃЌ
![]() (1)=
(1)= ![]() (3) +
(3) + ![]() (2)
(2)
![]() (3)=
(3)= ![]() (1)-
(1)- ![]() (2)
(2)
= [-393.51-(-282.98)]kJЁЄmol-1
= -110.53kJЁЄmol-1
ЗжЮіЂй,Ђк,ЂлШ§ИіЗНГЬЕФЙиЯЕПЩжЊЃКЗНГЬЂйМѕШЅЗНГЬЂкЕУЕНЗНГЬЂлЃЌЖј
![]() (3)=
(3)= ![]() (1)-
(1)- ![]() (2)
(2)
гЩДЫЃЌЮвУЧПЩвдЕУГіНсТлЃКЖрИіЛЏбЇЗДгІМЦСПЪНЯрМг(ЛђЯрМѕ)ЃЌЫљЕУЛЏбЇЗДгІМЦСПЪНЕФ ![]() (T)ЕШгкдИїМЦСПЪНЕФ
(T)ЕШгкдИїМЦСПЪНЕФ ![]() (T) ЯрМг(ЛђЯрМѕ)ЁЃ
(T) ЯрМг(ЛђЯрМѕ)ЁЃ
HessЖЈТЩЕФгІгУЃКгЩБъзМФІЖћЩњГЩьЪМЦЫу ![]()
![]()
![]() МЦЫуЙЋЪНЮЊЃК
МЦЫуЙЋЪНЮЊЃК
гЩДЫЃЌЮвУЧПЩвдЕУГіЃКдкЖЈЮТЖЈбЙЙ§ГЬжаЃЌЛЏбЇЗДгІЗДгІЕФБъзМФІЖћьЪБфЕШгкВњЮяЕФБъзМФІЖћЩњГЩьЪжЎКЭМѕШЅЗДгІЮяЕФБъзМФІЖћЩњГЩьЪжЎКЭЁЃ
HessЖЈТЩЕФгІгУ3ЃКгЩБъзМФІЖћШМЩеьЪМЦЫу ![]() (T)
(T)
МЦЫуЙЋЪНЮЊЃК
![]()
аЁНсЃЌдкБОПЮГЬжаЃЌЮвУЧЪзЯШЛиЙЫСЫИпжабЇЯАЕФжЊЪЖЃЌНщЩмСЫУшЪіЛЏбЇФмСПЙиЯЕЕФЯрЙиУћДЪЃЌв§ГіСЫаТЕФзДЬЌКЏЪ§ьЪЃЌвдМАЗДгІьЪБфЁЂБъзМФІЖћЩњГЩьЪЃЌЭЈЙ§ИЧЫЙЖЈТЩЧѓЕУЗДгІЙ§ГЬжаЕФьЪБфЁЃБОеТжаЕФЛЏбЇЗДгІЕФьЪБфМЦЫуЪЧжиЕуЃЌЧыЭЌбЇУЧНсКЯПЮКѓзївЕЃЌШЯецИДЯАЁЃ
ЕкЖўеТ ЛЏбЇЗДгІЕФЗНЯђЁЂЫйТЪКЭЯоЖШ
дкЛЏбЇШШСІбЇбаОПжаЃЌашвЊПМВьЮяРэБфЛЏКЭЛЏбЇБфЛЏЕФЕФЗНЯђадЁЃЪЕМЪЩЯЃЌздШЛНчжаШЮКЮздЗЂБфЛЏЙ§ГЬЖМЪЧОпгаЗНЯђадЕФЃЌЖјШШСІбЇЕквЛЖЈТЩЖдДЫВЂУЛгаИјГіШЮКЮЯожЦЁЃШШСІбЇЕкЖўЖЈТЩЧЁКУжИГіСЫетжжКъЙлБфЛЏЙ§ГЬНјааЕФЬѕМўКЭЗНЯђЁЃ
здЗЂБфЛЏЃЌМДздЗЂЙ§ГЬЃЈspontaneousprocessЃЉЕФЖЈвхЃКдквЛЖЈЬѕМўЯТВЛашвЊШЮКЮЭтСІЭЦЖЏОЭФмздЖЏНјааЕФЙ§ГЬЁЃШчЃКЫЎДгИпДІздЖЏСїЯђЕЭДІЃЌжБЕНЫЎЮЛВюЕШгкСуЪБЮЊжЙЃЌДЫЪБДяЕНСЫСІЦНКтЃЛШШДгИпЮТЮяЬхздЖЏЕиЯђЕЭЮТЮяЬхДЋЕнЃЌжБЕНЮТЖШВюЕШгкСуЪБЮЊжЙЃЌДяЕНШШЦНКтЃЛВЛЭЌХЈЖШЕФШмвКЛьКЯЪБЃЌШмжЪЛсздЖЏЕиДгИпХЈЖШЕФЕиЗНЭљЕЭХЈЖШЕФЕиЗНРЉЩЂЃЌжБЕНЬхЯЕИїВПЗжХЈЖШЯрЭЌЮЊжЙЃЌДяЕНЮяжЪЦНКтЃЛаПСЃЭЖШыЕНЙ§СПЕФСђЫсЭШмвКжаЛсздЖЏЕиЗЂЩњжУЛЛЗДгІЃЌжБЕНЛЏбЇЦНКтЮЊжЙЁЃЖјЫќУЧЕФФцЙ§ГЬЖМЪЧВЛПЩФмздЖЏНјааЕФЁЃ
здЗЂЙ§ГЬдкШШСІбЇжагжГЦЮЊВЛПЩФцЙ§ГЬ(irreversibleprocess)ЁЃЫљЮНВЛПЩФцЙ§ГЬОЭЪЧЮоТлгУЪВУДЗНЗЈЖМВЛФмЪЙЬхЯЕКЭЛЗОГЭЌЪБИДдЕФЙ§ГЬЃЌвВЪЧздШЛНчжаецЪЕДцдкЕФЙ§ГЬЁЃЖјПЩФцЙ§ГЬ(reversibleprocess)ЪЧЬхЯЕКЭЛЗОГПЩвдЭЌЪБИДдЕФЙ§ГЬЃЌЫќЪЧвЛжжШШСІбЇЩЯЕФЁЂРэЯыЛЏЕФздЗЂЙ§ГЬЃЌздШЛНчВЂВЛбЯИёДцдкЁЃ
здЗЂЙ§ГЬЕФЛљБОЬиеїЃК
ЃЈ1ЃЉЕЅЯђадЁЃЃЈ2ЃЉОпгазїЗЧЬхЛ§ЙІЕФБОСьЁЃЃЈ3ЃЉОпгавЛЖЈЕФЯоЖШЁЃ
ХаЖЯвЛИіЛЏбЇЗДгІФмЗёздЗЂЃЌЖдгкЛЏбЇбаОПКЭЛЏЙЄЩњВњОпгаживЊЕФвтвхЁЃвђЮЊЃЌШчЙћЪТЯШжЊЕРвЛИіЗДгІИљБОВЛПЩФмЗЂЩњЃЌШЫУЧОЭВЛБидйЛЈОЋСІШЅбаОПЫќЁЃ
ЭЦЖЏЛЏбЇЗДгІздЗЂЕФвђЫиЪЧЪВУДФиЃП
19ЪРМЭ70ФъДњЃЌЗЈЙњЕФБДШќТоЃЈBerthelotPEMЃЉКЭЕЄТѓЕФЬРФЗЫЙЃЈThomsonJЃЉОЭШЯЮЊЁАжЛгаЗХШШЗДгІВХФмздЗЂНјааЁБЁЃетжжЙлЕугавЛЖЈЕФЕРРэЃЌвђЮЊДІгкИпФмЬЌЕФЬхЯЕЪЧВЛЮШЖЈЕФЃЌЖјвЛАуРДЫЕЃЌЛЏбЇЗДгІЕФФмСПНЛЛЛвдШШЮЊжїЁЃОЙ§ЗДгІЃЌНЋвЛВПЗжФмСПвдШШЕФаЮЪНЪЭЗХИјЛЗОГЃЌЪЙИпФмЬЌЕФЗДгІЮяБфГЩЕЭФмЬЌЕФВњЮяЃЌЬхЯЕВХЛсИќЮШЖЈЁЃЪТЪЕЩЯЃЌЗХШШЗДгІвВЕФШЗДѓЖМЪЧздЗЂЗДгІЁЃгЩДЫПЩМћЃЌздШЛНчжаФмСПНЕЕЭЕФЧїЪЦЪЧЛЏбЇЗДгІздЗЂЕФвЛжжживЊЭЦЖЏСІЁЃ
ЪЧВЛЪЧЫљгаЮќШШЗДгІЖМВЛздЗЂФиЃПЪТЪЕВЂЗЧШчДЫЁЃ
Р§ШчЃЌДѓМвЫљЪьжЊЕФKNO3ШмгкЫЎЕФЙ§ГЬМШЪЧЮќШШЕФЃЌгжЪЧздЗЂЕФЁЃгжШчЬМЫсИЦЕФЗжНтЗДгІЃК
CaCO3(s)=CaO(s)+CO2(g)
дкИпЮТЃЈДѓдМ840ЁцвдЩЯЃЉвВЪЧздЗЂНјааЕФЃЌПЩЫќЪЧЮќШШЗДгІЁЃ
вђДЫЃЌФмСПЯТНЕЕФЧїЪЦВЂВЛЪЧЭЦЖЏЛЏбЇЗДгІздЗЂНјааЕФЮЈвЛвђЫиЁЃ
ПМВьЩЯУцЫљЪіЕФздЗЂНјааЖјгжЮќШШЕФЗДгІЛђЙ§ГЬПЩвдЗЂЯжЃЌЫќУЧгавЛИіЙВЭЌЕФЬиеїЃЌМДЙ§ГЬЛђЗДгІЗЂЩњКѓЬхЯЕЕФЛьТвГЬЖШдіДѓЁЃ
ШчKNO3ЙЬЬхжаK+РызгКЭNO3-РызгЕФХХВМЪЧЯрЖдгаађЕФЃЌЦфФкВПРызгЛљБОЩЯжЛдкОЇИёЕуеѓЩЯеёЖЏЁЃШмгкЫЎКѓЃЌK+КЭNO3-дкЫЎШмвКжавђЫќУЧЕФШШдЫЖЏЖјЪЙЛьТвГЬЖШДѓдіЁЃдйЦЉШчЃЌжаЙњгаОфГЩгяЃКЪЎФъГЩжЎВЛзуЃЌвЛЕЉЛйжЎгагрЁЃвВвўКЌгаЛьТвЖШШнвздіДѓжЎвтЁЃ
гЩДЫПЩМћЃЌЛьТвЖШдіДѓЕФЧїЪЦЪЧздЗЂЕФЛЏбЇЗДгІЕФгжвЛживЊЭЦЖЏСІЁЃ
дкетРяЃЌШЫУЧгУьиЃЈentropyЃЉРДДњБэЬхЯЕЛьТвЖШЕФДѓаЁЃЌГЃгУЗћКХSБэЪОЁЃЬхЯЕЕФЛьТвЖШдНДѓЃЌьижЕдНДѓЁЃШШСІбЇвбОжЄУїЃЌьиЪЧЬхЯЕЕФзДЬЌКЏЪ§ЁЃвђДЫЃЌьиБфЕФДѓаЁвВжЛШЁОігкЬхЯЕЕФЪМЬЌгыжеЬЌЃЌгыБфЛЏЭООЖЮоЙиЁЃьиБфЕФМЦЫуЙЋЪНвВвбгЩШШСІбЇЕМГіЃЈЦфЭЦЕМЙ§ГЬвбГЌГіБОНЬВФвЊЧѓЕФЗЖЮЇЃЌДЫДІДгТдЃЉЃЌМДЕБЬхЯЕЕФзДЬЌЗЂЩњБфЛЏЪБЃЌЦфьиБфЕШгкЬхЯЕгЩЪМЬЌжСжеЬЌОПЩФцЙ§ГЬетжжЭООЖБфЛЏЕФШШЮТЩЬЁЃьиЕФЕЅЮЛЪЧJЁЄK-1ЁЃашвЊМгвдЫЕУїЕФЪЧЃЌЬхЯЕгЩЭЌвЛЪМЬЌЕНЭЌвЛжеЬЌЃЌвВПЩОВЛПЩФцЙ§ГЬЕФЭООЖБфЛЏЙ§ШЅЃЌЕЋзДЬЌКЏЪ§ьиЕФБфЛЏжЕЁїSЪЧвЛбљЕФЁЃ
ьиЕФИХФюЪЧдк19ЪРМЭгЩПЫРЭаоЫЙЬсГіЕФЃЌЕЋгЩгкЕБЪБЖдетвЛИХФюШБЗІЮяРэвтвхЕФНтЪЭЃЌЙЪШЫУЧЖдьиГжЛГвЩКЭОмОјЕФЬЌЖШЁЃжБЕНВЃЖњзШТќЃЈL.E.BoltzmannЃЉАбьигыЬхЯЕзДЬЌЕФДцдкИХТЪСЊЯЕЦ№РДЃЌЪЙьигаСЫУїШЗЕФЮяРэвтвхЃЌВХЮЊШЫУЧЫљЙуЗКНгЪмЁЃ
жјУћЕФВЃЖњзШТќЙиЯЕЪНЮЊЃКS=kЉRІИ
ЪНжаkЮЊВЃЖњзШТќГЃЪ§ЃЌІИЮЊШШСІбЇИХТЪЃЌМДФГвЛКъЙлзДЬЌЫљЖдгІЕФЮЂЙлзДЬЌЪ§ЃЌІИдНДѓЃЌЛьТвЖШдНДѓЃЌьижЕSдНДѓЁЃетвЛЙиЯЕЪНЮЊКъЙлЮяРэСПЁЊьизїГіСЫЮЂЙлЕФНтЪЭЃЌНвЪОСЫШШЯжЯѓЕФБОжЪЃЌЕьЖЈСЫЭГМЦШШСІбЇЕФЛљДЁЃЌОпгаЛЎЪБДњЕФвтвхЁЃ
ьиSЪЧЬхЯЕЛьТвЖШЕФвЛжжСПЖШЁЃЖдгкШЮКЮДПЮяжЪЕФЭъећОЇЬхЃЈжИОЇЬхФкВПЮоШЮКЮШБЯн,жЪЕуХХСаЭъШЋгаађЃЌЮодгжЪЃЉРДЫЕЃЌдкОјЖдСуЖШЪБЃЌШШдЫЖЏМИКѕЭЃжЙЃЌЬхЯЕЕФЛьТвЖШзюЕЭЃЌЦфьижЕS0ЮЊСуЁЃЛђепЫЕ,ЁАШШСІбЇЮТЖШ0KЪБЃЌШЮКЮДПЮяжЪЕФЭъећОЇЬхЕФьижЕS0ЮЊСуЁБЃЌетОЭЪЧШШСІбЇЕкШ§ЖЈТЩ(the third law of thermodynamics)ЁЃвдДЫЮЊЯрЖдБъзМЧѓЕУЕФьижЕSTГЦЮЊЮяжЪЕФЙцЖЈьиЃЈconventional entropyЃЉЃЌвВНаОјЖдьиЁЃ
ВЂВЛЪЧЫљгаЮяжЪдкОјЖдСуЖШЪБЖМФмаЮГЩЭъећОЇЬх,ШчВЃСЇЬх,ЭЌЮЛЫиЙВДцЬх,ВЛЭъУРОЇЬх,жЪЕуХХСаЗНЪНЖржжЕФОЇЬхЕШ,ОЭВЛЪЧЭъећОЇЬх,ЦфОјЖдСуЖШЪБЕФьижЕвВВЛЮЊСу,МДЯрЖдЭъећОЇЬх,ЫќУЧОпгаЗЧСуЕФгаЯоьижЕ.ЕЋШШСІбЇПЩвджЄУї,етВЂВЛгАЯьЕкШ§ЖЈТЩЕФгІгУ,МДЙцЖЈьиИХФювВПЩвдгІгУгкЗЧЭъећОЇЬхЕФДПЮяжЪ.
дкБъзМзДЬЌЯТ1molДПЮяжЪдкЮТЖШTЪБЕФЙцЖЈьиГЦЮЊБъзМФІЖћЙцЖЈьи,МђГЦБъзМФІЖћьиЃЈstandard molar entropyЃЉ,гУSmӨ�������БэЪОЃЌЕЅЮЛЪЧJЁЄK-1ЁЄmol-1ЃЌвЛаЉЮяжЪSmӨ�������ЕФЪ§ОнМћЪщФЉИНБэЁЃвЊзЂвтЃЌЮШЖЈЕЅжЪЕФБъзМФІЖћьиВЛЮЊСуЃЌвђЮЊЫќУЧВЛЪЧОјЖдСуЖШЕФЭъећОЇЬхЁЃ
ашвЊжИГіЕФЪЧЃЌЫЎШмвКжаРызгЕФSmӨ�������ЃЌЪЧЙцЖЈдкБъзМзДЬЌЯТЫЎКЯHЃЋРызгЕФБъзМФІЖћьижЕЮЊСуЕФЛљДЁЩЯЧѓЕУЕФЯрЖджЕЁЃ
ИљОньиЕФвтвхЃЌЮяжЪЕФБъзМФІЖћьиSmӨ�������жЕвЛАуГЪЯжШчЯТЕФБфЛЏЙцТЩЃК
ЃЈ1ЃЉЭЌвЛЮяжЪЕФВЛЭЌОлМЏЬЌЃЌЦфSmӨ�������жЕЪЧЃК
SmӨ�������ЃЈЦјЬЌЃЉ>SmӨ�������ЃЈвКЬЌЃЉ>SmӨ�������ЃЈЙЬЬЌЃЉ
ЃЈ2ЃЉЖдгкЭЌвЛжжОлМЏЬЌЕФЭЌРраЭЗжзгЃЌИДдгЗжзгБШМђЕЅЗжзгЕФSmӨ�������жЕИќДѓЃЌШч
SmӨ�������ЃЈCH4,gЃЉ<SmӨ������� (C2H6,g)< SmӨ������� (C3H8,g)
ЃЈ3ЃЉЕБбЙЧПвЛЖЈЪБ,ЖдЭЌвЛОлМЏЬЌЕФЭЌжжЮяжЪЃЌЮТЖШЩ§ИпЃЌьижЕМгДѓЁЃ
ЃЈ4ЃЉдкЮТЖШвЛЖЈЪБ,ЖдЦјЬЌЮяжЪЃЌМгДѓбЙЧПЃЌьижЕМѕаЁ;ЖдЙЬЬЌКЭвКЬЌЮяжЪЃЌбЙЧПИФБфЖдЫќУЧЕФьижЕгАЯьВЛДѓЁЃ
ЯТУцЃЌЮвУЧРДМЦЫуЛЏбЇЗДгІьиБфЃКгЩБъзМФІЖћьиSmӨ������� (T)ПЩМЦЫуШЮвЛЛЏбЇЗДгІЕФБъзМьиБфЁїrSmӨ������� (T)ЃК
ЩшЛЏбЇЗДгІЮЊ:
aA+dD=eE+fF
дђЁїrSmӨ������� (T)=(eSӨ�������E+fSӨ�������F)ВњЮя-(aSӨ�������A+dSӨ�������D)ЗДгІЮя=ЁЦІдBSBӨ������� (B,T)
ІдBЮЊЯргІЮяжЪЕФЛЏбЇМЦСПЪ§ЁЃДЫЭт,гЩгкЮТЖШИФБфЪБЁЦSmӨ�������ЃЈВњЮяЃЉЕФИФБфКЭЁЦSmӨ�������ЃЈЗДгІЮяЃЉЕФИФБфЯрНќЃЌЖјЪщКѓИНТМжаБъзМФІЖћьиSmӨ�������ЪЧ298.15KЪБЕФЪ§ОнЃЌЙЪПЩвдНќЫЦШЯЮЊЃК
ЁїrSmӨ�������ЃЈTЃЉЁжЁїrSmӨ�������ЃЈ298.15KЃЉ
Р§ЬтЃК
ЬњЕФбѕЛЏЗДгІЮЊЃК4Fe(s)+3O2(g)=2Fe2O3(s)
ВщИНТМПЩжЊЃЌЯрЙиЮяжЪFe(s)ЁЂO2(g)ЁЂFe2O3(s)дк298.15KЯТЕФБъзМФІЖћьиSmӨ�������ЗжБ№ЮЊ27.28JЁЄmol-1ЁЄK-1ЃЌ205.14JЁЄmol-1ЁЄK-1ЃЌ87.40JЁЄmol-1ЁЄK-1ЁЃ
ЪдМЦЫудк298.15KЯТИУЗДгІЕФБъзМФІЖћьиБфЁїfSmӨ������� (298K)ЁЃ
НтЃКвђЮЊЁїfSmӨ������� (298K)=ЁЦІдBSBӨ������� (B,298K)ЙЪ
ЁїfSmӨ������� (298K)=2SmӨ������� [Fe2O3(s)]ЈC4SmӨ������� [Fe(s)]-3SmӨ������� [O2(g)]
=2ЁС87.40JЁЄmol-1ЁЄK-1ЈC4ЁС27.28JЁЄmol-1ЁЄK-1-3ЁС205.14JЁЄmol-1ЁЄK-1
=-549.74JЁЄmol-1ЁЄK-1
гЩздШЛНчжаЕФздЗЂБфЛЏПЩМћЃКШЮКЮЬхЯЕдкУЛгаЭтНчгАЯьЪБЃЌзмЪЧЕЅЯђЕиЧїгкФГжжМЋЯозДЬЌЁЊЁЊШШСІбЇЦНКтЬЌЃЌЖјОіВЛПЩФмздЖЏЕиФцЯђНјааЁЃетвЛНсТлОЭЪЧШШСІбЇЕкЖўЖЈТЩ(thesecondlawofthermodynamics)ЁЃ
дкШШСІбЇЬхЯЕжаЃЌЬхЯЕФмСПЕФБфЛЏЪЧЭЈЙ§гыЛЗОГНЛЛЛШШЛђЙІРДЪЕЯжЕФЁЃЖдгкгыЛЗОГжЎМфМШЮоЮяжЪЕФНЛЛЛвВЮоФмСПЕФНЛЛЛЕФЙТСЂЬхЯЕРДЫЕЃЌЭЦЖЏЬхЯЕФкЛЏбЇЗДгІздЗЂНјааЕФвђЫиОЭжЛгавЛИіЃЌФЧОЭЪЧьидіМгЁЃвђДЫ,в§ШыьиЕФИХФюжЎКѓ,ШШСІбЇЕкЖўЖЈТЩгжПЩБэЪіЮЊ:дкЙТСЂЬхЯЕФк,ШЮКЮБфЛЏЖМВЛПЩФмЪЙьиЕФзмжЕМѕЩйЁЃвВГЦьидіМгдРэЃЈprincipleofentropyincreaseЃЉЁЃЦфЪ§бЇБэДяЪНЮЊЃКЁїSЙТСЂЁн0
ЪНжаЁїSЙТСЂБэЪОЙТСЂЬхЯЕЕФьиБфЁЃШчЙћБфЛЏЙ§ГЬЪЧздЗЂЙ§ГЬ,дђЁїSЙТСЂ>0;ШчЙћЬхЯЕДІгкЦНКтзДЬЌ,дђЁїSЙТСЂ=0;змжЎьигадіЮоМѕЁЃ
ЪЕМЪЩЯ,ьидіМгдРэвВЪЧЙТСЂЬхЯЕжаЙ§ГЬФмЗёздЗЂНјааЛђЬхЯЕЪЧЗёДІгкЦНКтзДЬЌЕФХаЖЯвРОн,МђГЦьиХаОн.
еце§ЕФЙТСЂЬхЯЕЪЧВЛДцдкЕФЃЌвђЮЊЬхЯЕКЭЛЗОГжЎМфзмЛсДцдкЛђЖрЛђЩйЕФФмСПНЛЛЛЁЃШчЙћЮвУЧАбгыЬхЯЕгаЯрЛЅзїгУЕФФЧвЛВПЗжЛЗОГвВАќРЈНјШЅЃЌОЭЙЙГЩСЫвЛИіаТЕФЬхЯЕЃЌетИіаТЬхЯЕПЩвдПДГЩЪЧвЛИіДѓЕФЙТСЂЬхЯЕЃЌЦфьиБфЮЊЁїSзмЮЊЃКЁїSзм=ЁїSЬхЯЕ+ЁїSЛЗОГ
ЁїSзмЃО0здЗЂБфЛЏ
ЁїSзмЃМ0ЗЧздЗЂБфЛЏ
ЁїSзм=0ЦНКтзДЬЌ
РћгУИУЪНЮвУЧПЩвдНтОіЛЏбЇЗДгІздЗЂНјааЗНЯђЕФХаЖЯЮЪЬтЁЃжЛвЊЫуГіЬхЯЕЕФьиБфКЭЛЗОГЕФьиБфЕФСНепжЎКЭМДПЩЃЌЕЋгІгУЦ№РДКмВЛЗНБуЃЌМШвЊМЦЫуЬхЯЕЕФьиБфгжвЊМЦЫуЛЗОГЕФьиБфЁЃЖдгкЛЗОГьиБфЕФМЦЫуЃЌгаЪБЪЧКмИДдгЕФЃЌгаЪБЛЙВЛКУМЦЫуЁЃ
ЖдгкФГаЉЛЏбЇЗДгІЃЌЬхЯЕЕФьиБфЁїSЬхЯЕе§КУЪЧЗДгІЕФьиБфЁЃЃЌШШСІбЇбаОПНсЙћБэУїЃЌЖдгкЖЈЮТЖЈбЙЯТЛЗОГЕФьиБфе§БШгкЗДгІЕФьЪБфЁїrHЕФИКжЕЃЌЗДБШгкЛЗОГЕФШШСІбЇЮТЖШЃЈОјЖдЮТЖШЃЉЃЌМД
ЁїSЛЗОГ=-![]()
ЫфШЛЃЌдкетРяЖдДЫЪНВЛФмЭЦЕМЃЌЕЋШдШЛФмЙЛБШНЯаЮЯѓЕиЫЕУїжЎЁЃвђЮЊЬхЯЕдкЖЈбЙЬѕМўЯТЗДгІЃЌШєЗХШШЗДгІЃЌдђЬхЯЕИјгшЛЗОГЕФШШЕШгкЗДгІьЪБфЁїHЬхЯЕЃЌЖјЖдгкЛЗОГРДЫЕЃЌЯрЕБгкЛЗОГвдПЩФцЙ§ГЬДгЬхЯЕЮќЪеЕФШШQrЛЗОГЃЌЫљвдQrЛЗОГ=-ЁїHЬхЯЕЃЌжЎЫљвдЫЕЛЗОГДЫЪБНќЫЦгкПЩФцЙ§ГЬЃЌЪЧгЩгкДѓЛЗОГДгЗДгІЮќЪеЕФШШНіНів§Ц№ЮЂаЁЕФБфЛЏЁЃЙЪЃЌШдПЩвдШЯЮЊЛЗОГБЃГжКуЮТЃЌЛЗОГЕФьиБфОЭПЩвджБНггУЦфПЩФцЙ§ГЬЕФШШЮТЩЬРДМЦЫуЁЃ
дкХаЖЯЛЏбЇЗДгІздЗЂадКЭЗНЯђЪБЃЌШчЙћФмЙЛевЕНвЛИіМДВЛгУПМТЧЛЗОГвђЫиЃЌжЛашПМТЧЬхЯЕБОЩэЕФЧщПіЃЌОЭПЩвдНтОіДѓЖрЪ§ЛЏбЇЗДгІЕФздЗЂадКЭЗНЯђЮЪЬтЕФХаОнЃЌНЋИќЮЊЗНБуЁЃЮЊДЫЃЌгаБивЊв§ШыаТЕФШШСІбЇКЏЪ§ЃЌвдзїЮЊдкЯргІЬѕМўЯТЃЌЙ§ГЬздЗЂЛђЗДгІздЗЂЕФЗНЯђМАЯоЖШЕФХаЖЯвРОнЁЃЖјДѓЖрЪ§ЛЏбЇЗДгІЖМЪЧдкЖЈЮТЖЈбЙЬѕМўЯТНјааЕФ,ЧвздЗЂЕФЛЏбЇЗДгІгжЪєгкШШСІбЇЩЯЕФздЗЂЙ§ГЬЁЃдкЖЈЮТЖЈбЙЯТЁїSзм=ЁїSЬхЯЕ+ЁїSЛЗОГ=ЁїSЬхЯЕ-![]() =ЁїS-
=ЁїS-![]() вдШШСІбЇЮТЖШTЭЌЪБГЫвдЕШЪНСНБпЃЌЕУ
вдШШСІбЇЮТЖШTЭЌЪБГЫвдЕШЪНСНБпЃЌЕУ
TЁїSзм=TЁїS-ЁїHСюЁїG=-TЁїS
ЁїG=ЁїH-TЁїSИУЪНБЛГЦЮЊGibbsЃHelmholtzЙЋЪНЁЃ
GibbsКЏЪ§БЛЖЈвхЮЊЃКG=HЈCTSGгжБЛГЦЮЊGibbsздгЩФмЁЃЁїGЮЊGibbsКЏЪ§БфЛђGibbsздгЩФмБфЁЃGгЩзДЬЌКЏЪ§HЁЂTЁЂSзщКЯЖјГЩЃЌЙЪзщКЯжЕGвВЪЧЬхЯЕЕФзДЬЌКЏЪ§ЃЌВЂЧвгыHгаЯрЭЌЕФЕЅЮЛЁЃИљОнЁїGгыЁїSзмЕФЙиЯЕЃЌПЩгЩьиХаОнЕУЕНGibbsКЏЪ§ХаОнЛђGibbsздгЩФмХаОнЃКдкВЛзіЗЧЬхЛ§ЙІКЭЖЈЮТЖЈбЙЯТЃЌШЮКЮздЗЂБфЛЏзмЪЧЬхЯЕЕФGibbsздгЩФмМѕЩйЁЃМД
ЁїSзмЃО0ЃЌЁїGT,PЃМ0ЗДгІе§ЯђздЗЂЃЈВЛПЩФцЙ§ГЬЃЉ
ЁїSзм=0ЃЌЁїGT,P=0ЗДгІЬхЯЕДІгкЦНКтзДЬЌЃЈПЩФцЙ§ГЬЃЉ
ЁїSзмЃМ0ЃЌЁїGT,PЃО0ЗДгІе§ЯђВЛздЗЂЃЈВЛздЗЂЙ§ГЬЃЉ
ИљОнGibbsздгЩФмЕФЖЈвхЃЌG=H-TSЃЌдђдкЖЈбЙЖЈЮТЬѕМўЯТЃЌзДЬЌБфЛЏЕФздгЩФмБфЁїGT,PЮЊЃК
ЁїGT,P=ЁїH-Ёї(TS)=ЁїH-TЁїS-SЁїT=ЁїH-TЁїS
МДЁїGT,P=ЁїHT-TЁїST
ЩЯЪНОЭЪЧжјУћЕФGibbsЃHelmholtzЙЋЪНЁЃДЫЙЋЪНАбгАЯьЙ§ГЬздЗЂЛђЛЏбЇЗДгІздЗЂЕФСНИівђЫиЃКФмСПБфЛЏЃЈетРяБэЯжЮЊКубЙЙ§ГЬШШЛђКубЙЗДгІШШЁїHЃЉгыЛьТвЖШБфЛЏСПЖШЃЈМДЙ§ГЬьиБфЛђЗДгІьиБфЁїSЃЉЭъУРЕиЭГвЛЦ№РДСЫЁЃДгGibbsЃHelmholtzЙЋЪНПЩвдПДГіЃЌЮТЖШTЖдМЊВМЫЙздгЩФмБфЁїGвВгаУїЯдгАЯьЁЃЯрЖдЖјбдЃЌВЛЩйБфЛЏЙ§ГЬЕФЁїHКЭЁїSЫцЮТЖШБфЛЏЕФИФБфжЕКмаЁЃЌЙЪвЛАуПЩВЛПМТЧЮТЖШTЖдЁїHКЭЁїSЕФгАЯьЃЌЕЋВЛФмКіТдЮТЖШTЖдЁїGЕФгАЯьЁЃ
дкВЛЭЌЮТЖШЯТЙ§ГЬздЗЂЛђЗДгІздЗЂНјааЕФЗНЯђШЁОігкЁїHКЭTЁїSжЕЕФЯрЖдДѓаЁЁЃЯжЗжБ№ЬжТлШчЯТЃЈЮЊСЫМђБуЃЌдкДЫЬжТлжаЪЁТдСЫШШСІбЇКЏЪ§ЕФЯТБъБэЪОЃЉЃК
ИљОнGibbsКЏЪ§ХаОнЃЌЁїGЁм0ЃЌМДЁїH-TЁїSЁм0ЗДгІе§ЯђздЗЂЛђДяЕНЦНКтЁЃдђЃК
(1)ЁїHЃО0ЃЌЁїSЃО0ЃЌМДЗХШШЁЂьидіМгЕФЙ§ГЬЛђЗДгІЃЌАДGibbsЃHelmholtzЙЋЪНЃЌдкШЮКЮЮТЖШЯТОљгаЁїGЃМ0ЃЌМДШЮКЮЮТЖШЯТЙ§ГЬЛђЗДгІЖМПЩФме§ЯђздЗЂЁЃ
(2)ЁїHЃО0ЃЌЁїSЃМ0ЃЌМДЮќШШЁЂьиМѕаЁЕФЙ§ГЬЛђЗДгІЃЌгЩгкСНИівђЫиЖМЖдЗДгІздЗЂНјаа
ВЛРћЃЌАДGibbsЃHelmholtzЙЋЪНЃЌдкШЮКЮЮТЖШЖМгаЁїGЃО0ЃЌДЫРрЧщПіе§ЯђзмЪЧВЛПЩФмздЗЂНјааЁЃ
(3)ЁїHЃМ0ЃЌЁїSЃМ0ЃЌМДЗХШШЁЂьиМѕаЁЕФЙ§ГЬЛђЗДгІЃЌАДЪНGibbsЃHelmholtzЙЋЪНЃЌЕЭ
ЮТгаРћгкЙ§ГЬЛђЗДгІе§ЯђздЗЂНјааЁЃ
(4)ЁїHЃО0ЃЌЁїSЃО0ЃЌМДЮќШШЁЂьидіМгЕФЙ§ГЬЛђЗДгІЃЌАДGibbsЃHelmholtzЙЋЪНЃЌИпЮТгаРћгкЙ§ГЬЛђЗДгІе§ЯђздЗЂНјааЁЃ
ДгЩЯУцЕФЗжЮіПЩвдПДГіЃЌЕБЁїHКЭЁїSетСНИігАЯьЗДгІздЗЂадЕФвђЫиЖМгаРћгкЗДгІздЗЂНјааЃЌЛђЖМВЛРћгкЗДгІЕФздЗЂНјааЪБЃЌЦѓЭМЭЈЙ§ЕїНкЮТЖШРДИФБфЗДгІздЗЂадЕФЗНЯђЪЧВЛПЩФмЕФЁЃжЛгаЁїHКЭЁїSетСНИівђЫиЖдздЗЂадЕФгАЯьЯрЗДЃЈЖўепе§ЁЂИКЗћКХЯрЭЌЕФЧщПіЃЉЃЌМДвЛИігаРћЃЌСэвЛИіВЛРћЪБЃЌВХПЩФмЭЈЙ§ИФБфЮТЖШЃЌРДИФБфЗДгІздЗЂНјааЕФЗНЯђЃЌЖјЁїG=0ЪБЕФЮТЖШЃЌМДЛЏбЇЗДгІДяЕНЦНКтЪБЕФЮТЖШЃЌвВНаГЦЮЊзЊБфЮТЖШЃК
TзЊБф=![]()
дкЗХШШьиМѕЕФЧщПіЯТЃЌетИіЮТЖШЪЧЗДгІФме§ЯђНјааЕФзюИпЮТЖШЃЛдкЮќШШьидіЕФЧщПіЯТЃЌетИіЮТЖШЪЧЗДгІФме§ЯђНјааЕФзюЕЭЮТЖШЁЃ
ЖдгкШЮвтвЛЖЈбЙЖЈЮТВЛзіЗЧЬхЛ§ЙІЕФЛЏбЇЗДгІЃК
aA+dD=eE+fF
ИљОнGibbsЃHelmholtzЙЋЪНЃЌИУЗДгІЕФGibbsздгЩФмБфЮЊ
ЁїrGT,P=ЁїrH(T)-TЁїrS(T)
ШєЖЈбЙЬѕМўЮЊБъзМбЙЧПPӨ�������ЃЌдђЪЧЗДгІЕФБъзМздгЩФмБфЃЌМДЃК
ЁїrGmӨ�������ЃЈTЃЉ=ЁїrHmӨ������� (T)-TЁїrSmӨ������� (T)
ЦфжаЁїrHmӨ������� (T)=(eЁїfHEӨ�������+fЁїfHFӨ�������)ВњЮя-(aЁїfHAӨ�������+dЁїfHDӨ�������)ЗДгІЮя
ЁїrSmӨ������� (T)=(eSEӨ�������+fSFӨ�������)ВњЮя-(aSAӨ�������+dSDӨ�������)ЗДгІЮя
вђДЫЃЌжЛвЊФмЙЛМЦЫуГіЁїrGӨ�������mЃЈTЃЉЕФНсЙћЃЌБуПЩвдХаЖЯБъзМЬЌЯТЛЏбЇЗДгІздЗЂНјааЕФЗНЯђЁЃ
Р§ЬтЃКвбжЊЁїfHmӨ������� [C6H6(l),298K]=49.10kJЁЄmol-1ЃЌЁїfHmӨ������� [C2H2(g),298K]=226.73kJЁЄmol-1ЃЛ
SmӨ������� [C6H6(l),298K]=173.40JЁЄmol-1K-1ЃЌSmӨ������� [C2H2(g),298K]=200.94JЁЄmol-1K-1ЁЃЪдХаЖЯЃК
ЗДгІC6H6(l)=3C2H2(g)дк298.15KЃЌБъзМЬЌЯТе§ЯђФмЗёздЗЂЃПВЂЙРЫузюЕЭЗДгІЮТЖШЁЃ
НтЃКИљОнGibbsЃHelmholtzЙЋЪН
ЁїrGmӨ�������ЃЈTЃЉ=ЁїrHmӨ������� (T)-TЁїrSmӨ������� (T)
ЁїrGmӨ�������ЃЈ298KЃЉ=ЁїrHmӨ������� (298K)-TЁїrSӨ�������m(298K)
ЖјЁїrHmӨ������� (298K)=3ЁїrHmӨ������� [C2H2(g),298K]ЃЁїrHmӨ������� [C6H6(l),298K]
=3ЁС226.73kJЁЄmol-1Ѓ1ЁС49.10kJЁЄmol-1
=631.09kJЁЄmol-1
ЁїrSmӨ������� (298K)=3SmӨ������� [C2H2(g),298K]ЃSmӨ������� [C6H6(l),298K]
=3ЁС200.94JЁЄmol-1K-1Ѓ1ЁС173.40JЁЄmol-1K-1
=429.42JЁЄmol-1K-1
ЙЪЁїrGmӨ�������ЃЈ298KЃЉ=631.09kJЁЄmol-1Ѓ298.15KЁС429.42ЁС10-3kJЁЄmol-1K-1
=503.06kJЁЄmol-1ЃО0е§ЯђЗДгІВЛздЗЂЁЃ
ШєЪЙЁїrGmӨ�������ЃЈTЃЉ=ЁїrHӨ�������m(T)-TЁїrSӨ�������m(T)ЃМ0ЃЌдђе§ЯђздЗЂЁЃ
гжвђЮЊЁїrHӨ�������mЁЂЁїrSӨ�������mЫцЮТЖШБфЛЏВЛДѓЃЌМД
ЁїrGӨ�������mЃЈTЃЉЁжЁїrHӨ�������m(298K)-TЁїrSӨ�������m(298K)ЃМ0
дђЃЌTЃО631.09kJЁЄmol-1/429.42ЁС10-3kJЁЄmol-1K-1=1469.6K
ЙЪзюЕЭЗДгІЮТЖШЮЊ1469.6KЁЃ
гУGibbsЃHelmholtzЙЋЪНМЦЫуЛЏбЇЗДгІЕФБъзММЊВМЫЙздгЩФмБфЁїrGmӨ�������ЃЌЩцМАЕНКубЙЗДгІШШКЭЗДгІьиБфЕФМЦЫуЃЌЗЧГЃЗГЫіЁЃШчЙћШЫУЧАбвЛаЉЬиЪтЗДгІЕФЁїrGmӨ�������ЕФвбжЊЪ§ОнСаГЩБэЃЌВЩгУгыЗДгІШШЧѓЫуЯрРрЫЦЕФЗНЗЈЃЌОЭПЩвдИќМгМђБуЕиМЦЫуШЮвЛЛЏбЇЗДгІЕФздгЩФмБфЁЃ
гЩгкGЪЧзДЬЌКЏЪ§ЃЌЛЏбЇЗДгІЕФЁїGвВгІИУжЛШЁОігкЪМЁЂжеЬЌЃЌгыЫљОРњЕФЭООЖЮоЙиЁЃвђДЫЃЌЦфGibbsздгЩФмБфгІЮЊЃК
ЁїrG=ЁЦGЃЈВњЮяЃЉ-ЁЦGЃЈЗДгІЮяЃЉ
ДгG=H-TSПДЃЌЫфШЛЮвУЧПЩвдЧѓГіЗДгІЮТЖШTЯТЕФЙцЖЈьиSЃЌЕЋЮвУЧЮоЗЈжЊЕРHЕФОјЖджЕЃЌвђДЫвВЮоЗЈШЗЖЈздгЩФмGЕФОјЖджЕЁЃвЊМЦЫуЗДгІЕФЁїrG,ОЭЕУгУРрЫЦгЩБъзМЩњГЩьЪМЦЫуЗДгІШШЕФЗНЗЈНтОіЁЃ
гЩзюЮШЖЈЕЅжЪЩњГЩ1molЮяжЪЕФЩњГЩЗДгІЕФGibbsздгЩФмБфГЦЮЊИУЮяжЪЕФФІЖћЩњГЩGibbsКЏЪ§ЁЃдкБъзМзДЬЌЯТФГЮяжЪЕФФІЖћЩњГЩGibbsКЏЪ§ГЦЮЊИУЮяжЪЕФБъзМФІЖћЩњГЩGibbsКЏЪ§ЛђБъзМФІЖћЩњГЩздгЩФмЃЈstandardmolarfreeenergyofformationЃЉ,ЗћКХЁїfGmӨ�������ЃЌЕЅЮЛЮЊkJЁЄmol-1ЁЃ
АДееБъзМФІЖћЩњГЩздгЩФмЁїfGmӨ�������ЕФЖЈвхЃЌШШСІбЇЪЕМЪЩЯвбЙцЖЈЮШЖЈЕЅжЪЕФБъзМФІЖћЩњГЩздгЩФмЮЊСуЁЃЯёЁїfHmӨ�������вЛбљЃЌЁїfGmӨ�������вВЪЧЯрЖджЕЁЃИїжжЮяжЪЕФЁїfGmӨ�������Ъ§ОнМћЪщФЉИНТМЁЃ
ШШСІбЇПЩжЄУїЃЌРћгУЪщФЉИНБэжаВщЕНЕФЁїfGmӨ�������жЕЃЈБэжавЛАуЪЧ289.15KЕФЪ§ОнЃЉЃЌАДееИЧЫЙЖЈТЩЧѓЗДгІШШЕФЗНЪНЃЌЭЈЙ§МгЁЂМѕЕФЗНЗЈПЩвдЧѓЫудк298.15KЁЂВЛзіЗЧЬхЛ§ЙІЬѕМўЯТЕФШЮвЛЛЏбЇЗДгІЕФБъзМздгЩФмБфЁїrGmӨ�������ЁЃШчЛЏбЇЗДгІЮЊЃК
aA+dD=eE+fF
дђЁїrGӨ�������m(298K)=(eЁїfGӨ�������E+fЁїfGӨ�������F)ВњЮя-(aЁїfGAӨ�������+dЁїfGDӨ�������)ЗДгІЮя
=ЁЦІдBЁїfGBӨ������� (298K)
зЂвтЃЌгыЁїfHmӨ�������КЭЁїfSmӨ�������ВЛЭЌЃЌЮТЖШЖдЁїfGmӨ�������гаКмДѓгАЯьЁЃ
ЯТУцЮвУЧОйР§ЫЕУїЃК
вбжЊдк298.15KЪБNH3(g)ЕФБъзМФІЖћЩњГЩздгЩФмЁїfGmӨ�������ЮЊ-16.45kJЁЄmol-1ЃЌМЦЫуКЯГЩАБЗДгІЃКN2(g)+3H2(g)=2NH3(g)ЕФЁїrGmӨ�������ЃЈ298KЃЉЁЃ
НтЃКИљОнКЯГЩАБЕФЗДгІЗНГЬЪН
ЁїrGmӨ�������ЃЈ298KЃЉ=2ЁїfGmӨ������� [NH3(g)]Ѓ[1ЁСЁїfGmӨ������� [N2(g)]+3ЁїfGmӨ������� [H2(g)]
=2ЁС(-16.45kJЁЄmol-1)Ѓ0kJЁЄmol-1
=-32.90kJЁЄmol-1
ЁїrGmӨ�������жЛПЩвдХаЖЯБъзМЬЌЯТЛЏбЇЗДгІздЗЂНјааЕФЗНЯђЁЃЪЕМЪгІгУжаЃЌЗДгІЛьКЯЮяКмЩйДІгкЯргІЕФБъзМзДЬЌЁЃЗДгІНјааЪБЃЌЦјЬхЮяжЪЕФЗжбЙКЭШмвКжаШмжЪЕФХЈЖШОљдкВЛЖЯБфЛЏжЎжаЃЌжБжСЦНКтЃЌЁїrGm=0ЁЃЁїrGmВЛНігыЮТЖШгаЙиЃЌЖјЧвгыЬхЯЕзщГЩгаЙиЁЃ
ЭЈЙ§ШШСІбЇПЩЕМГіЛЏбЇЗДгІдкЖЈбЙЖЈЮТЃЈЗЧБъзМЬЌЃЉЬѕМўЯТGibbsздгЩФмБфЁїrGmЕФМЦЫуЙЋЪНЃЌМДЁїrGmгыЬхЯЕзщГЩжЎМфЕФЙиЯЕЃК
ЁїrGT,P=ЁїrGTӨ�������+RT1nJ
ДЫЪНГЦЮЊЛЏбЇЗДгІЕШЮТЗНГЬЪНЁЃЪНжаЁїrGT,PЪЧЮТЖШЮЊTЕФЗЧБъзМЬЌЯТЗДгІЕФGibbsздгЩФмБфЃЌJЮЊЗДгІЩЬЁЃЫљЮНЗЧБъзМЬЌЃЌЪЧжИЖЈбЙЖЈЮТЬѕМўжаВЮгыЗДгІЕФЦјЬхЕФЫВЪБЗжбЙВЛЪЧБъзМбЙЧПpӨ�������ЃЌВЮгыЗДгІЁЂДІгкШмвКзДЬЌЕФЮяжжЕФЫВЪБХЈЖШВЛЪЧБъзМХЈЖШcӨ�������(1molЁЄL-1)ЁЃЁїrGTӨ�������ЪЧИУЗДгІдкБъзМЬЌЯТФІЖћздгЩФмБфЃЌМДДЫЪБВЮгыЗДгІЕФЦјЬхЕФЫВЪБЗжбЙЮЊБъзМбЙЧПpӨ�������ЃЌВЮгыЗДгІЁЂДІгкШмвКзДЬЌЕФЮяжжЕФЫВЪБХЈЖШЮЊБъзМХЈЖШcӨ�������ЃЛRЪЧЦјЬхГЃЪ§ЃЌTЪЧШШСІбЇЮТЖШЁЃ
![]() ШєЛЏбЇЗДгІЮЊaA(aq)+dD(s)=eE(aq)+fF(g)ЪєЖрЯрЗДгІЃЌЧвЦјЯрЮЊРэЯыЦјЬхЃЌКіТдбЙЧПЖдвКЁЂЙЬЯрЕФгАЯьЁЃдђЗДгІЩЬJЕФБэДяЪНЮЊЃК
ШєЛЏбЇЗДгІЮЊaA(aq)+dD(s)=eE(aq)+fF(g)ЪєЖрЯрЗДгІЃЌЧвЦјЯрЮЊРэЯыЦјЬхЃЌКіТдбЙЧПЖдвКЁЂЙЬЯрЕФгАЯьЁЃдђЗДгІЩЬJЕФБэДяЪНЮЊЃК
J=
ЦфжаВЮгыЗДгІЕФЦјЬхЮяжЪвдЯрЖдЗжбЙБэЪОЃЛВЮгыЗДгІЁЂДІгкШмвКзДЬЌЕФЮяжЪвдЯрЖдХЈЖШБэЪОЃЛВЮгыЗДгІЕФДПвКЬхКЭДПЙЬЬхЮяжЪЃЌЦфЯрЖдХЈЖШЕШгк1ЛђепЛюЖШa=1ЃЌдђВЛБиаДШыЗДгІЩЬJЕФБэДяЪНжаЁЃ
![]() ШчЖрЯрЗДгІZn(s)+2HЃЋ(aq)=Zn2ЃЋ(aq)+H2(g)ЃЌЦфЗДгІЩЬJЕФБэДяЪНЮЊЃК
ШчЖрЯрЗДгІZn(s)+2HЃЋ(aq)=Zn2ЃЋ(aq)+H2(g)ЃЌЦфЗДгІЩЬJЕФБэДяЪНЮЊЃК
J=
ЭЈГЃЃЌЛЏбЇЗДгІЖМОпгаПЩФцадЁЃЕБШЛЃЌгааЉЛЏбЇЗДгІМИКѕФмНјааЕНЕзЃЌШчТШЫсМиЗжНтЗДгІЃЌдкMnO2ДпЛЏЯТKClO3ЛљБОЩЯФмШЋВПзЊБфЮЊKClКЭO2ЁЃгжШчЃЌЗХЩфаддЊЫиЕФЭЩБфЁЂТШгыЧтЛђбѕгыЧтЕФБЌеЈЪНЗДгІЕШЃЌетаЉЗДгІГЦЮЊВЛПЩФцЗДгІЁЃЪЕМЪЩЯОјДѓЖрЪ§ЛЏбЇЗДгІЖМЪЧВЛФмНјааЕНЕзЕФЗДгІЃЌвВОЭЪЧПЩФцЗДгІ(reversiblereaction)ЁЃПЩФцЗДгІВЂЗЧШШСІбЇжаЕФПЩФцЙ§ГЬЃЌЦфе§ЯђЗДгІЪЧвЛИіздЗЂЙ§ГЬЃЌЦфФцЯђЗДгІвВЪЧвЛИіздЗЂЙ§ГЬЁЃжЛЪЧВЛЭЌЕФЗДгІЃЌЦфПЩФцГЬЖШВЛЭЌЖјвбЁЃ
ЮвУЧАбдквЛЖЈЬѕМўЃЈЮТЖШЁЂбЙЧПЁЂХЈЖШЕШЃЉЯТЃЌЕБе§ЗДСНИіЗНЯђЕФЗДгІЫйТЪЯрЕШЪБЃЌЗДгІЮяКЭВњЮяЕФХЈЖШВЛдйЫцЪБМфЖјБфЛЏЕФзДЬЌЃЌГЦЮЊЛЏбЇЦНКт(chemicalequillibrium)ЃЌетвВОЭЪЧЛЏбЇЗДгІЫљФмДяЕНЕФзюДѓЯоЖШЁЃжЛвЊЭтНчЬѕМўВЛБфЃЌетИізДЬЌОЭВЛдйЫцЪБМфЖјБфЛЏЃЌЕЋЭтНчЬѕМўвЛЕЉИФБфЃЌЦНКтзДЬЌОЭвЊЗЂЩњБфЛЏЁЃЦНКтзДЬЌДгКъЙлЩЯПДЫЦКѕЪЧОВжЙЕФЃЌЕЋЪЕМЪЩЯетВЂВЛвтЮЖзХЗДгІвбОЭЃжЙЃЌДгЮЂЙлЩЯПДЪЧвЛжжЖЏЬЌЦНКтЁЃДЫЭтЃЌЛЙгІЫЕУїЕФЪЧЃЌЛЏбЇЦНКтЪЧжИдЪМЕФЗДгІЮяКЭзюКѓВњЮяжЎМфДяГЩЕФЦНКтЃЌЫќгыЗДгІЕФвЛВНЛђЗжМИВННјааЮоЙиЁЃ
дквЛЖЈЬѕМўЯТВЛЭЌЕФЛЏбЇЗДгІНјааЕФГЬЖШЪЧВЛЯрЭЌЕФЃЌЖјЧвЭЌвЛЗДгІдкВЛЭЌЕФЬѕМўЯТЃЌЫќНјааЕФГЬЖШвВгаКмДѓЕФВюБ№ЁЃдкИјЖЈЬѕМўЯТЃЌШчКЮПижЦЗДгІЬѕМўДгЖјШЗЖЈЗДгІНјааЕФзюДѓЯоЖШЃПетаЉЖМЪЧЛЏбЇЦНКтЫљбаОПЕФЮЪЬтЁЃЛЏбЇЦНКтВЛНідкНтЪЭаэЖрДПЛЏбЇаджЪЕФЙ§ГЬКЭЗДгІжаЪЧживЊЕФЃЌЖјЧвдкНтЪЭАќРЈбЊвКЁЂЬхвККЭЯИАћЮяжЪЕФЩњУќЬхЯЕвдМАЯйЗжУкЕФЙ§ГЬЕШвВЪЧживЊЕФЁЃ
ПЩФцЗДгІЕФР§згКмЖрЃЌШчЃК
CO+H2OЁЊЁЊCO2+H2
Sn2+(aq)+2Fe3+(aq)ЁЊЁЊSn4+(aq)+2Fe2+(aq)
ПЩФцЗДгІДІгкЛЏбЇЦНКтзДЬЌЪБЃЌОпгаШчЯТЛљБОЬиеїЃК
ЃЈ1ЃЉЯЕЭГЕФзщГЩВЛдйЫцЪБМфЖјБфЁЃ
ЃЈ2ЃЉЛЏбЇЦНКтЪЧЖЏЬЌЦНКтЁЃ
ЃЈ3ЃЉЦНКтзщГЩгыДяЕНЦНКтЕФЭООЖЮоЙиЁЃ
ЖдШЮвтвЛПЩФцЕФЛЏбЇЗДгІaA+dD=eE+fFЃЌЦфЗЧБъзМЬЌЯТGibbsздгЩФмБфЁїrGmПЩгУЛЏбЇЗДгІЕШЮТЗНГЬЪНМЦЫуЃК
ЁїrGT,P=ЁїrGTӨ�������+RT1nJ
ЕБЗДгІДяЕНЦНКтЪБЃЌЗДгІЕФздгЩФмБфЁїrGTЃЌP=0ЃЌДЫЪБЗДгІЮяКЭВњЮяЕФХЈЖШЛђЗжбЙЮЊЦНКтХЈЖШЛђЦНКтЗжбЙЃЌВЛдйЫцЪБМфБфЛЏЃЌМДКъЙлЩЯЗДгІВЛдйМЬајНјааЃЌетвВОЭЪЧЛЏбЇЗДгІЕФЯоЖШЁЃДЫЪБЕФЗДгІЩЬJЃЌМДЮЊБъзМЦНКтГЃЪ§ЃЈstandard equilibrium constantЃЉЃЌгУЗћКХKӨ�������БэЪОЃЌДњШыЩЯЪНЃЌЕУЃК
0=ЁїrGTӨ�������+RT1nKӨ�������
ЙЪЃЌЁїrGTӨ�������=-RT1nKӨ�������
ДЫЪНвВОЭЪЧгЩБъзМGibbsздгЩФмБфМЦЫуЛЏбЇЗДгІЕФБъзМЦНКтГЃЪ§ЕФЙЋЪНЁЃдкЭЌвЛЛЏбЇЗДгІжаЃЌБъзМЦНКтГЃЪ§KӨ�������ЕФБэДяЪНгыЗДгІЩЬJЕФБэДяЪНЯрЭЌЃЌжЛЪЧБъзМЦНКтГЃЪ§KӨ�������ЕФБэДяЪНжаcAЁЂcDЛђЁЂPAЁЂPDКЭcEЁЂcFЁЂЛђPEЁЂPFЗжБ№БэЪОЗДгІЮяКЭВњЮяЕФЦНКтХЈЖШЛђЦНКтЗжбЙЃЛЖјЗДгІЩЬJЕФБэДяЪНжаcAЁЂcDЛђЁЂPAЁЂPDКЭcEЁЂcFЁЂЛђPEЁЂPFЗжБ№БэЪОЗДгІЮяКЭВњЮяЕФЫВЪБХЈЖШЛђЫВЪБЗжбЙЁЃ
ШчЖрЯрЗДгІZn(s)+2HЃЋ(aq)=Zn2ЃЋ(aq)+H2(g)ЃЌЦфБъзМЦНКтГЃЪ§KӨ�������ЕФБэДяЪНЮЊЃК
KӨ�������=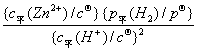
БъзМЦНКтГЃЪ§KӨ�������гыЮТЖШгаЙиЃЌгыХЈЖШЛђЗжбЙЮоЙиЁЃKӨ�������ЪЧвЛИіЮоСПИйЕФЮяРэСПЁЃ
ДЫЭтЃЌБъзМЦНКтГЃЪ§ЕФЪ§жЕКЭБъзМЦНКтГЃЪ§ЕФБэДяЪНЖМгыЛЏбЇЗДгІЗНГЬЪНЕФаДЗЈгаЙиЁЃШчКЯГЩАБЕФЗДгІЃК
N2(g)+3H2(g)=2NH3(g)
KӨ�������1=![]()
![]() N2(g)+
N2(g)+![]() H2(g)DNH3(g)
H2(g)DNH3(g)
KӨ�������2=![]()
дкЮТЖШЯрЭЌЪБЃЌKӨ�������1КЭKӨ�������2ЕФЪ§жЕВЛвЛбљЃЌСНепжЎМфЕФЙиЯЕЮЊK1Ө�������=ЃЈK2Ө�������ЃЉ2ЁЃЮЊЗНБуЦ№МћЃЌБъзМЦНКтГЃЪ§KӨ�������ЕФаДЗЈШдНЋбигУЯАЙпадЕФЦНКтГЃЪ§KЕФБэДяЃЌЕЋЪЕМЪЩЯЦНКтГЃЪ§KЕФЪ§ОнЖМЪЧБъзМЦНКтГЃЪ§KӨ�������ЕФЪ§ОнЃЌЬиДЫЫЕУїЁЃ
Р§ЬтЃКЧѓ298.15KЪБЗДгІ2SO2(g)+O2(g)D2SO3(g)ЕФБъзМЦНКтГЃЪ§KӨ�������ЁЃвбжЊЁїfGmӨ������� (SO2) = -300.2 KJЁЄmol-1ЃЌЁїfGmӨ������� (SO3)=-371.1KJЁЄmol-1ЁЃ
НтЃКИУЗДгІЕФЁїrGmӨ�������ЮЊЃК
ЁїrGmӨ�������=2ЁїfGmӨ������� (SO3)-2ЁїfGmӨ������� (SO2)-ЁїfGmӨ������� (O2)
=2ЁС(-371.1KJЁЄmol-1)-2ЁС(-300.2KJЁЄmol-1)ЈC0KJЁЄmol-1
=-141.8KJЁЄmol-1
ЖјЃЌЁїrGmӨ�������=-RT1nKӨ�������
ЙЪЃЌ1nKӨ�������=-ЁїrGӨ�������m/RT
=![]() =57.2
=57.2
KӨ�������=7.0ЁС1024
ШЗЖЈБъзМЦНКтГЃЪ§Ъ§жЕЕФзюЛљБОЗНЗЈЪЧЪЕбщВтЖЈЃЌжЛвЊВтЖЈЪЕбщЦНКтГЃЪ§ЃЌОЭПЩЭЈЙ§ЫќгыБъзМЦНКтГЃЪ§ЕФЙиЯЕМЦЫуЕУЕНЁЃШчЫФбѕЛЏЖўЕЊЕФЗжНтЗДгІФЫЕфаЭЕФПЩФцЗДгІЃК
N2O4(g)Ёњ2NO2(g)
ЪЕбщВтЖЈБэУїЃЌдк373KЁЂЗДгІДяЦНКтЪБЃЌNO2гыN2O4ЕФЮяжЪЕФСПХЈЖШАДЯТЪНЧѓГіЕФБШжЕЮЊвЛГЃЪ§ЃЌМДЃК
K=![]() =0.36
=0.36
ЪНжаKГЦЮЊЪЕбщЦНКтГЃЪ§ЃЈexperimental equilibrium constantЃЉЁЃЕБВЮгыЗДгІЕФЮяжЪдкЪНжаЕФХЈЖШЯюЛђЗжбЙЯюжБНггУХЈЖШЃЈвдЮяжЪЕФСПХЈЖШЮЊЕЅЮЛЃЉЛђЗжбЙЃЈвдkPaЮЊЕЅЮЛЃЉБэЪОЪБЃЌЦфЪЕбщЦНКтГЃЪ§БэДяЪНгыБъзМЦНКтГЃЪ§ЕФБэДяЪНЛљБОЯрЭЌЃЈЪ§жЕЩЯПЩФмгаЫљВЛЭЌЃЉЃЛЮЈвЛЕФВЛЭЌдкгкБъзМЦНКтГЃЪ§БэДяЪНжаЕФХЈЖШЯюЛђЗжбЙЯюЪЧгУЯрЖдХЈЖШЛђЯрЖдЗжбЙРДБэЪОЕФЁЃЪЕбщЦНКтГЃЪ§KcКЭKpБэДяЪНЕФЪщаДддђвВДѓЬхгыБъзМЦНКтГЃЪ§KӨ�������ЕФЯрЭЌЁЃ
ЖдгкШЮвтвЛШмвКжаЕФЗДгІЃК
aA+dD=eE+fF
ЪЕбщВтЖЈБэУїЃЌЕБЗДгІДяЕНЦНКтЪБЃЌШєЗДгІЮяЕФЦНКтХЈЖШЮЊ[A]ЁЂ[D]КЭВњЮяЕФЦНКтХЈЖШЮЊ[E]ЁЂ[F]ЃЌдђЫќУЧжЎМфЕФЙиЯЕПЩгУЯТЪНБэЪОЃК
Kc=![]()
ЪНжаKcГЦЮЊХЈЖШЦНКтГЃЪ§ЃЛЗДгІЮяКЭВњЮяЕФЦНКтХЈЖШНдЮЊЮяжЪЕФСПХЈЖШЁЃШчЙћa+d=e+fЃЌдђKcЮоЕЅЮЛЃЛШчЙћa+dЁйe+fЃЌдђKcгаЕЅЮЛЃЌЦфЕЅЮЛЮЊЃЈmolЁЄL-1ЃЉЃЈe+fЃЉ-ЃЈa+dЃЉЁЃЦфЪЕЃЌВЛТлKcгаЮоЕЅЮЛЃЌKӨ�������КЭKcдкЪ§жЕЩЯЪЧЯрЕШЕФЁЃ
ШєЩЯЪіЗДгІЮЊЦјЯрЗДгІЃЌдђЃК
Kp=![]()
ЪНжаЃЌpЃЈAЃЉЁЂpЃЈDЃЉКЭpЃЈEЃЉЁЂpЃЈFЃЉЗжБ№ЮЊЗДгІЮяКЭВњЮяЕФЦНКтЗжбЙЃЌЕЅЮЛЮЊkPaЃЌKpГЦЮЊЗжбЙЦНКтГЃЪ§ЁЃЕБa+d=e+fЪБЃЌKpЮоЕЅЮЛЃЌKpдкЪ§жЕЩЯЕШгкKӨ�������ЃЛЕБa+dЁйe+fЪБ,KpгаЕЅЮЛЃЌKpЕФЪ§жЕВЛЕШгкKӨ�������ЁЃШєЗДгІЮяКЭВњЮяЕФЦНКтЗжбЙЃЌЕЅЮЛЮЊБъзМДѓЦјбЙЃЌдђKpвВГЦЮЊЗжбЙЦНКтГЃЪ§ЁЃЕБa+d=e+fЪБЃЌKpЮоЕЅЮЛЃЌKpдкЪ§жЕЩЯЕШгкKӨ�������ЃЛЕБa+dЁйe+fЪБ,KpгаЕЅЮЛЃЌKpЕФЪ§жЕШдШЛЕШгкKӨ�������ЁЃKcКЭKpОљГЦЮЊЪЕбщЦНКтГЃЪ§ЁЃЪЕбщЦНКтГЃЪ§жЕдНДѓЃЌЛЏбЇЗДгІе§ЯђНјааЕФГЬЖШдНГЙЕзЃЌетвЛЕугыKӨ�������ЪЧЯрЭЌЕФЁЃЪЕбщЦНКтГЃЪ§дкЛЏбЇЦНКтЕФМЦЫужаШддкЙуЗКгІгУЁЃ
ЯТУцЮвУЧОйР§бЇЯАKӨ�������ЕФгІгУЃК
дквЛЖЈЬѕМўЯТЃЌЛЏбЇЗДгІДяЕНЦНКтЪБЃЌе§ЁЂЗДСНИіЗНЯђЕФЗДгІЫйТЪЯрЕШЃЌЦНКтзщГЩЃЈЗДгІЮяКЭВњЮяЕФХЈЖШЃЉВЛдйЫцЪБМфЖјБфЛЏЁЃетБэУїЗДгІЮяЯђВњЮязЊБфДяЕНСЫзюДѓЯоЖШЁЃ
KӨ�������ЕФЪ§жЕЗДгГСЫЛЏбЇЗДгІЕФБОадЃЌKӨ�������жЕдНДѓЃЌе§ЯђЗДгІПЩФмНјааЕФГЬЖШдНДѓЃЛKӨ�������жЕдНаЁЃЌе§ЯђЗДгІНјааЕУдНВЛЭъШЋЁЃвђДЫЃЌБъзМЦНКтГЃЪ§KӨ�������ЪЧвЛЖЈЮТЖШЯТЃЌЛЏбЇЗДгІПЩФмНјааЕФзюДѓЯоЖШЕФСПЖШЁЃ
ЗДгІНјааЕФГЬЖШвВГЃгУЦНКтзЊЛЏТЪРДБэЪОЁЃAЮяжЪЕФЦНКтзЊЛЏТЪІСЃЈAЃЉБЛЖЈвхЮЊЃК
![]()
ЪНжаЮЊn0ЃЈAЃЉЗДгІПЊЪМЪБAЕФЮяжЪЕФСПЃЛneqЃЈAЃЉЮЊЦНКтЪБAЕФЮяжЪЕФСПЁЃKӨ�������дНДѓЃЌЭљЭљІСЃЈAЃЉвВдНДѓЁЃ
НЋЁїrGӨ�������T=-RT1nKӨ�������ДњШыЁїrGT,P=ЁїrGӨ�������T+RT1nJжаЃЌПЩЕУЃК
ЁїrGT,P=-RT1nKӨ�������+RT1nJ
ДЫЪНЪЧЛЏбЇЗДгІЕШЮТЗНГЬЪНЕФСэвЛБэДяаЮЪНЁЃДгЩЯЪНПЩжЊЃЌБШНЯБъзМЦНКтГЃЪ§KӨ�������гыЗДгІЩЬJЕФЯрЖдДѓаЁЃЌПЩвдХаЖЯЖЈбЙЖЈЮТЁЂВЛзіЗЧЬхЛ§ЙІЪБЃЌЛЏбЇЗДгІздЗЂНјааЕФЗНЯђЃК
ЕБJЃМKӨ�������ЪБЃЌдђЁїrGT,PЃМ0ЃЌе§ЯђЗДгІздЗЂЃЛ
ЕБJ=KӨ�������ЪБЃЌдђЁїrGT,P=0ЃЌЛЏбЇЗДгІДяЕНЦНКтЃЛ
ЕБJЃОKӨ�������ЪБЃЌдђЁїrGT,PЃО0ЃЌФцЯђЗДгІздЗЂЁЃ
етвВЪЧдЄВтПЩФцЗДгІЗНЯђЕФживЊХаОнЁЃШчЙћЗДгІЩЬJВЛЕШгкБъзМЦНКтГЃЪ§KӨ�������ЃЌОЭБэУїЗДгІЬхЯЕДІгкЗЧЦНКтЬЌЃЌДЫЪБЬхЯЕОпгаДге§ЯђЛђФцЯђздЖЏЕиГЏзХЦНКтЬЌдЫЖЏЕФЧїЪЦЁЃЖдгкЛЏбЇЗДгІРДЫЕЃЌОЭЪЧДцдкздЗЂНјааЗДгІЕФЧїЪЦЁЃJжЕгыKӨ�������жЕЯрВюдНДѓЃЌДге§ЯђЛђФцЯђздЗЂНјааЗДгІЕФЧїЪЦОЭдНДѓЁЃ
СэЭтЃЌШєвбжЊЗДгІЬхЯЕЕФЦ№ЪМзщГЩЃЌРћгУKӨ�������ПЩМЦЫуЦНКтЪБЗДгІЬхЯЕЕФзщГЩЁЃ
Р§ЬтЃКвбжЊЗДгІCO(g)+Cl2(g)=COCl2(g)
дкЖЈЮТКуШнЬѕМўЯТНјаа,373KЪБKӨ�������=1.5ЁС108ЁЃ
ЗДгІПЊЪМЪБc0(CO)=0.0350molЁЄL-1ЃЌc0(Cl2)=0.0270molЁЄL-1ЃЌc0(COCl2)=0ЁЃМЦЫу373KЗДгІДяЕНЦНКтЪБИїЮяжжЕФЗжбЙКЭCOЕФЦНКтзЊЛЏТЪЁЃ
НтЃКpV=nRTвђЮЊTЁЂVВЛБфЃЌpЁиnBp=cRT
p0(CO)=(0.0350ЁС8.314ЁС373)kPa=106.3kPa
p0(Cl2)=(0.0270ЁС8.314ЁС373)kPa=82.0kPa
CO(g)+Cl2(g)=COCl2(g)
ПЊЪМcB/(molЁЄL-1) 0.035 00.0270 0
ПЊЪМpB/kPa 106.3 82.0 0
МйЩшCl2ШЋВПзЊЛЏЃЌ106.3-82.0 0 82.0
гжЩшCOCl2зЊЛЏ x x -x
ЦНКтpB/kPa 24.3+x x 82.0-x
KӨ�������=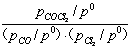 =
=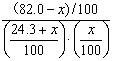 =1.5ЁС108
=1.5ЁС108
вђЮЊKКмДѓЃЌxКмаЁЃЌМйЩш82.0-xЁж82.0ЃЌ24.3+xЁж24.3ЁЃ
![]() =1.5ЁС108x=2.3ЁС10-6
=1.5ЁС108x=2.3ЁС10-6
ЦНКтЪБЃКp(CO)=24.3kPaЃЌp(Cl2)=2.3ЁС10-6kPaЃЌp(COCl2)=82.0kPa
![]()
=![]()
=77.1%
ЛЏбЇЦНКтЖМЪЧЯрЖдЕФЃЌднЪБЕФЁЃЕБЭтНчЬѕМўИФБфЪБЃЌЦНКтОЭЛсБЛЦЦЛЕЃЌИїжжЮяжЪЕФХЈЖШЃЈЛђЗжбЙЃЉвВЛсИФБфЃЌЗДгІНЋМЬајНјааЃЌжБЕНдкаТЕФЬѕМўЯТНЈСЂаТЕФЦНКтЁЃетжжгЩгкЬѕМўБфЛЏЕМжТЛЏбЇЦНКтвЦЖЏЕФЙ§ГЬЃЌГЦЮЊЛЏбЇЦНКтЕФвЦЖЏЃЈshiftofchemicalequilibriumЃЉЁЃ
ЖдгкШЮвтвЛЛЏбЇЗДгІЃЌЦфЖЈбЙЖЈЮТЯТЕФGibbsздгЩФмБфЮЊЃК
ЁїrGT,P=-RT1nKӨ�������+RT1nJ
ШчЙћЗДгІЩЬгыБъзМЦНКтГЃЪ§ЕФЙиЯЕЮЊЃКJ=KӨ�������ЃЌдђЁїrGT,P=0ЃЌЗДгІДяЕНЦНКтЁЃЕБдіМгЗДгІЮяЕФХЈЖШЛђМѕЩйВњЮяЕФХЈЖШЪБЃЌНЋЪЙJ<KӨ�������ЃЌдђЁїrGT,P<0ЃЌНЋДђЦЦдгаЦНКтЃЌе§ЯђЗДгІНЋздЗЂНјааЃЌжБЕНдйвЛДЮЪЙJ=KӨ�������ЃЌЗДгІНЈСЂаТЕФЦНКтЮЊжЙЁЃЗДжЎЃЌШчЙћдіМгВњЮяЕФХЈЖШЛђМѕЩйЗДгІЮяЕФХЈЖШЃЌНЋЕМжТJ>KӨ�������ЃЌЁїrGT,P>0ЃЌФцЯђЗДгІНЋздЗЂНјааЃЌжБжСаТЕФЦНКтЁЃ
бЙСІЖдвКЯрКЭЙЬЯрЗДгІЕФЦНКтМИКѕУЛгагАЯьЃЌЕЋЖдгкЦјЬхВЮгыЕФШЮвтвЛЛЏбЇЗДгІЃК
aA+dDЁњeE+fF
діМгЗДгІЮяЕФЗжбЙЛђМѕЩйВњЮяЕФЗжбЙЃЌЖМНЋЪЙJ<KӨ�������ЃЌЁїrGT,P<0ЃЌЦНКтЯђгввЦЖЏЁЃЗДжЎЃЌдіДѓВњЮяЕФЗжбЙЛђМѕЩйЗДгІЮяЕФЗжбЙЃЌНЋЪЙJ>KӨ�������ЃЌЁїrGT,P>0ЃЌЦНКтЯђзѓвЦЖЏЁЃетгыХЈЖШЖдЛЏбЇЦНКтЕФгАЯьЭъШЋЯрЭЌЁЃ
ЖдгквЛИівбДяЦНКтЕФЦјЯрЗДгІЃЌШчЙћдіМгЛђМѕаЁЬхЯЕЕФзмбЙЃЌНЋЗжвдЯТСНжжЧщПіЖдЛЏбЇЦНКтВњЩњгАЯьЃК
ЂйЕБa+d=e+fЪБ,МДЗДгІЮяЦјЬхЗжзгзмЪ§гыВњЮяЦјЬхЗжзгзмЪ§ЯрЕШЃЌдђдіМгзмбЙгыНЕЕЭзмбЙЖМНЋВЛЛсИФБфJжЕЃЌШдШЛЮЌГжJ=KӨ�������,ЛЏбЇЦНКтНЋВЛЗЂЩњвЦЖЏЃЛ
ЂкШчЙћЗДгІЮяЦјЬхЗжзгзмЪ§гыВњЮяЦјЬхЗжзгзмЪ§ВЛЯрЕШЃЌМДa+dЁйe+fЃЌИФБфзмбЙНЋИФБфJжЕЃЌЪЙJЁйKӨ�������ЃЌЦНКтНЋЗЂЩњвЦЖЏЁЃдіМгзмбЙСІЃЌЦНКтНЋЯђЦјЬхЗжзгзмЪ§МѕЩйЕФЗНЯђвЦЖЏЁЃМѕаЁзмбЙСІЃЌЦНКтНЋЯђЦјЬхЗжзгзмЪ§діМгЕФЗНЯђвЦЖЏЁЃ
ХЈЖШЛђбЙСІЖдЛЏбЇЦНКтЕФгАЯьжЛИФБфJжЕЃЌЖјВЛИФБфБъзМЦНКтГЃЪ§KӨ�������ЁЃЖјЮТЖШЖдЛЏбЇЦНКтЕФгАЯьШДЭъШЋВЛЭЌЃЌвђЮЊгЩБъзМздгЩФмБфМЦЫуЛЏбЇЗДгІЕФБъзМЦНКтГЃЪ§ЕФЙЋЪНПЩжЊЃЌЮТЖШИФБфЃЌKӨ�������жЕвВНЋЗЂЩњИФБфЁЃМДЃК
ЁїrGmӨ�������=-RT1nKӨ�������ЃЌгжвђЮЊЁїrGmӨ�������=ЁїrHmӨ�������ЈCTЁїrSmӨ�������
ЙЪНЋСНЪНКЯВЂЃЌПЩЕУЃК
1nKӨ�������=-![]() +
+![]()
ЩшдкЮТЖШЮЊT1КЭT2ЪБЗДгІЕФБъзМЦНКтГЃЪ§ЗжБ№ЮЊK1Ө�������КЭK2Ө�������ЃЌВЂМйЖЈЮТЖШЖдЁїrHmӨ�������КЭЁїrSmӨ�������ЕФгАЯьПЩвдКіТдЃЌдђ
ЃЈ1ЃЉ1nK1Ө�������=-![]() +
+![]()
ЃЈ2ЃЉ1nK2Ө�������=-![]() +
+![]()
гЩЃЈ2ЃЉ-ЃЈ1ЃЉЕУЃК
ln![]() =
=![]()
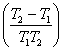
ЩЯЪНБэЪОСЫБъзМЦНКтГЃЪ§KӨ�������гыЮТЖШЕФЙиЯЕЁЃЭЈЙ§ВтЖЈВЛЭЌЮТЖШTЯТЕФKӨ�������жЕЃЌгУ1nKӨ�������Жд1/TзїЭМПЩЕУвЛжБЯпЃЌгЩжБЯпаБТЪКЭНиОрПЩвдЧѓЕУЛЏбЇЗДгІЕФЁїrHmӨ�������КЭЁїrSmӨ�������ЁЃ
ДгЩЯЪНПЩЬНЬжЮТЖШЖдЛЏбЇЦНКтЕФгАЯьЁЃЖдгке§ЯђЮќШШЗДгІЃЌЁїrHmӨ�������>0ЃЌЕБЩ§ИпЮТЖШЪБЃЌМДT2>T1ЃЌБиШЛгаK2Ө�������>K1Ө�������ЃЌЦНКтНЋе§ЯђвЦЖЏЃЛЖдгке§ЯђЗХШШЗДгІЃЌЁїrHmӨ�������<0ЃЌЕБЩ§ИпЮТЖШЪБЃЌМДT2>T1ЃЌдђБигаK2Ө�������<K1Ө�������ЃЌОЭЪЧЫЕЦНКтНЋЯђФцЗДгІЗНЯђвЦЖЏЁЃ
злЩЯЃЌХЈЖШЁЂбЙСІЁЂЮТЖШЕШвђЫиЖдЛЏбЇЦНКтЕФгАЯьЃЌПЩвдгУ1884ФъЗЈЙњЛЏбЇМвРеЯФЬиСаЃЈLeChatelierЃЉзмНсГіЕФвЛЬѕЦеБщЙцТЩРДХаЖЯЃКЦНКтзмЪЧЯђзХЯћГ§ЭтРДгАЯьЃЌЛжИДдгазДЬЌЕФЗНЯђвЦЖЏЁЃетОЭЪЧжјУћЕФРеЯФЬиСаЦНКтвЦЖЏдРэЁЃИУдРэЪЪгУгкШЮКЮвбДяГЩЦНКтЕФЬхЯЕЃЌЮяРэЦНКтЕФЬхЯЕврВЛР§ЭтЁЃУЛгаДяГЩЦНКтЕФЬхЯЕЃЌВЛФмгІгУРеЯФЬиСадРэЁЃ
жСДЫЃЌЮвУЧбЇЭъЛЏбЇЗДгІЦНКтЕФХаЖЯЃЌЧыЭЌбЇУЧНсКЯЯАЬтЃЌИДЯАЫљбЇжЊЪЖЃЌЫМПМВЛЭЌЧщПіЯТЃЌЛЏбЇЗДгІНјааЕФЗНЯђЮЪЬтЁЃ
ЕкШ§еТ ЫсМюЗДгІКЭГСЕэЗДгІ
ШЫУЧЖдЫсМюЕФШЯЪЖОРњСЫвЛИігЩЧГШыЩюЃЌгЩЕЭМЖЕНИпМЖЕФШЯЪЖЙ§ГЬЁЃзюГѕЕФжБЙлШЯЪЖЃКЫсЃКгаЫсЮЖЃЌФмЪЙЪЏШяЪдвКБфКьЕФЃЌШчЪГДзЁЃМюЃКгаЩЌЮЖЃЌЛЌФхИаЃЌФмЪЙКьЩЋЪЏШяБфРЖЃЌВЂФмгыЫсЗДгІЩњГЩбЮКЭЫЎЃЌШчаЁЫеДђЁЃ
1884ФъЃЌШ№ЕфЛЏбЇМвArrheniusИљОнЕчНтжЪЛЏбЇРэТлЃЌЖдЫсМюзїСЫвдЯТЖЈвхЃК
ЫсЃКдкЫЎШмвКжаОЕчРыжЛЩњГЩH+вЛжжбєРызгЁЃ
МюЃКдкЫЎШмвКжаОЕчРыжЛЩњГЩOH-вЛжжвѕРызгЁЃ
ИУЖЈвхЪЙШЫРрЖдЫсМюЕФШЯЪЖЪЕЯжСЫДгЯжЯѓЕНБОжЪЕФЗЩдОЃЌЕЋИУЖЈвхвВгаОжЯоадЃЌЫќАбЫсКЭМюжЛЯогкЫЎШмвКЁЃЫцзХШЫУЧЖдЫсМюШЯЪЖЕФРЉеЙЁЃШЫУЧЯрМЬЬсГіСЫШмМСРэТлЃЌжЪзгРэТлЃЌЕчзгРэТлКЭШэгВЫсМюЕФРэТлЁЃБОеТНЋвдЫсМюжЪзгРэТлЮЊжааФЃЌЬжТлЫсМюЦНКтЮЪЬтЁЃ
Ыс:ЗВЪЧФмЪЭЗХжЪзгH+ЕФШЮКЮКЌЧтдзгЕФЗжзгЛђРызгЕФЮяжжЃЌМДжЪзгЕФИјгшЬхЁЃ
МюЃКШЮКЮФмгыжЪзгНсКЯЕФЗжзгЛђРызгЕФЮяжжЁЃМДжЪзгЕФНгЪмЬхЁЃ
![]()
![]()
ПЩМћЃЌЫсИјГіжЪзгЩњГЩЯргІЕФМюЃЌЖјМюНсКЯжЪзгКѓгжЩњГЩЯргІЕФЫсЃЛЫсгыМюжЎМфЕФетжжвРРЕЙиЯЕГЦЙВщюЙиЯЕЁЃЯргІЕФвЛЖдЫсМюБЛГЦЮЊЙВщюЫсМюЖдЁЃР§ШчЃКHAcЕФЙВщюМюМюЪЧAc-ЃЌAc-ЕФЙВщюЫсЪЧHAcЃЌHAcКЭAc-ЪЧвЛЖдЙВщюЫсМюЁЃ
МШФмИјГіжЪзгЃЌЖдФмНгЪмжЪзгЕФЮяжЪЮЊСНадЮяжЪЃЌР§ШчЃК
HPO42-,H2PO42-,[Fe(OH)(H2O)5]2+ЃЌH2OЕШЁЃ
ИљОнЫсМюжЪзгРэТлЃКЫсМюНтРыЗДгІЪЧжЪзгзЊвЦЗДгІЁЃ
Р§ШчЃКHFдкЫЎШмвКжаЕФНтРыЗДгІЪЧгЩИјГіЕФжЪзгЕФАыЗДгІКЭНгЪмжЪзгЕФАыЗДгІзщГЩЕФЁЃ
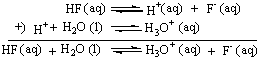
ЫЎЪЧСНадЮяжЪЃЌЫќЕФздЩэНтРыЗДгІвВЪЧжЪзгзЊвЦЗДгІЁЃ
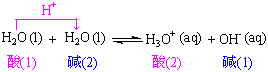
бЮРрЫЎНтЗДгІвВЪЧРызгЫсМюЕФжЪзгзЊвЦЗДгІЁЃР§ШчЃКNaAcЕФЗжНтЃК
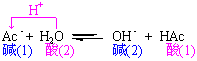
NH4+ЕФЗжНтЃК
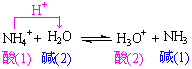
ЫсМюжаКЭЗДгІЃЈАќРЈЗЧЫЎШмМСжаЕФЗДгІЃЉвВЪЧжЪзгзЊвЦЗДгІЁЃ
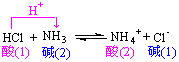
![]()
ЫсКЭМюЕФЧПЖШЪЧжИЫсИјГіжЪзгЕФФмСІКЭМюНгЪмжЪзгЕФФмСІЕФЧПШѕЁЃ
дкЫЎШмвКжаЃК![]()
гЩДЫПЩвдЫЕУїдкЫЎШмвКжаЃЌHAcЕФЫсадБШHCNЕФЫсадЧПЁЃ
ЧјЗжаЇгІЃКгУвЛИіШмМСАбЫсЛђМюЕФЯрЖдЧПШѕЧјЗжПЊРДЃЌГЦЮЊШмМСЕФЁАЧјЗжаЇгІЁБЁЃР§ШчЃКH2OПЩвдЧјЗжHAcЁЂHCNЫсадЕФЧПШѕЁЃ
РЦНаЇгІЃКШмМСНЋЫсЛђМюЕФЧПЖШРЦНЕФзїгУЃЌГЦЮЊШмМСЕФЁАРЦНаЇгІЁБЁЃЫЎЖдЧПЫсЦ№ВЛЕНЧјЗжзїгУЃЌЫЎФмЭЌЕШЧПЖШЕиНЋHClO4ЃЌHClЃЌHNO3ЕШЧПЫсЕФжЪзгШЋВПељШЁЙ§РДЁЃ
бЁШЁБШЫЎМюадШѕЕФМюЃЌШчБљДзЫсЮЊШмМСЖдЫЎжаЕФЧПЫсПЩЬхЯжГіЧјЗжаЇгІЁЃР§ШчЩЯЪіЧПЫсдкБљДзЫсжаВЛЭъШЋНтРыЃЌЫсадЧПЖШвРДЮЮЊЃК HClO4>HCl>H2SO4>HNO3ЃЌЫљвдH2OЖдвдЩЯЧПЫсгаРЦНЗДгІЃЌБљДзЫсЖдЫћУЧгаЧјЗжаЇгІЁЃ
НсТлЃКЫсаддНЧПЃЌЦфЙВщюМюдНШѕЃЛМюдНЧПЃЌЦфЙВщюЫсдНШѕЁЃ
ЫсадЃКHClO4>H2SO4>H3PO4>HAc>H2CO3>NH4+>H2O
МюадЃКClO4-<HSO4-<H2PO4-<Ac-<HCO3-<NH3<OH-
АДееЫсМюжЪзгРэТлЃЌЫЎЕФздЩэНтРыЦНКтПЩБэЪОЮЊЃК
![]() Лђ
Лђ![]()
БъзМЦНКтГЃЪ§БэДяЪНЃК
![]()
Лђ
![]()
![]() ЁЊЁЊЫЎЕФРызгЛ§ГЃЪ§ЃЌМђГЦЫЎЕФРызгЛ§ЁЃ
ЁЊЁЊЫЎЕФРызгЛ§ГЃЪ§ЃЌМђГЦЫЎЕФРызгЛ§ЁЃ
25ЁцЪБЃЌДПЫЎжаc(H+)=c(OH-)=1.0ЁС10-7molЁЄL-1
![]() =1.0ЁС10-14
=1.0ЁС10-14
100ЁцЪБЃЌДПЫЎ![]() =5.43ЁС10-13
=5.43ЁС10-13
ПЩМћЃЌЫцзХЮТЖШЩ§ИпЃЌЫЎЕФ![]() діДѓЁЃ
діДѓЁЃ
ШмвКжаH3O+ХЈЖШЛђOH-ХЈЖШЕФДѓаЁЗДгГСЫШмвКЕФЕФЫсМюадЕФЧПШѕЁЃвЛАуЯЁШмвКжаЃЌc(H3O+)ЕФХЈЖШЗЖЮЇдкЃЈ10-1~10-14ЃЉmolЁЄL-1жЎМфЁЃдкЛЏбЇПЦбЇжаЃЌЭЈГЃЯАЙпгквдc(H3O+)ЕФИКЖдЪ§РДБэЪОЦфКмаЁЕФЪ§СПМЖЁЃ
МДpH=-{lgc(H3O+)}ЃЛгыpHЖдгІЕФЛЙгаpOHЃЌМДpOH=-lg{c(OH-)}
дк25ЁцЪБЃЌ
![]() =1.0ЁС10-14
=1.0ЁС10-14
![]() =14.00
=14.00
![]()
![]()
pHЪЧгУРДБэЪОЫЎШмвКЫсМюадЕФвЛжжБъЖШЁЃpHгњаЁЃЌc(H3O+)гњДѓЃЌШмвКЕФЫсадгњЧПЁЃ
ЭЈГЃЫљЫЕЕФШѕЫсКЭШѕМюЪЧжИЫсЁЂМюЕФЛљБОДцдкаЮЪНЮЊжаадЗжзгЁЃЫќУЧДѓВПЗжвдЗжзгаЮЪНДцдкгкШмвКжаЃЌжЛгаЩйВПЗжгыЫЎЗЂЩњжЪзгзЊвЦЗДгІЁЃжЛФмИјГівЛИіжЪзгЕФГЦЮЊвЛдЊШѕЫсЃЌФмИјГіЖрИіжЪзгЕФЮЊЖрдЊШѕЫсЃЛжЛФмНгЪмвЛИіжЪзгЕФЮЊвЛдЊШѕМюЃЌФмНгЪмЖрИіжЪзгЮЊЖрдЊШѕМюЁЃ
дквЛдЊШѕЫсHAcЕФЫЎШмвКжаЁЃДцдкзХЯТСажЪзгзЊвЦЗДгІЃК
![]()
ЦфЦНКтГЃЪ§
![]() ЁЊШѕЫсЕФHAЕФНтРыГЃЪ§ЁЃШѕЫсЕФНтРыГЃЪ§ПЩвдНшжњpHМЦВтЖЈШмвКЕФpHРДШЗЖЈЁЃШєвбжЊШѕЫсЕФНтРыГЃЪ§
ЁЊШѕЫсЕФHAЕФНтРыГЃЪ§ЁЃШѕЫсЕФНтРыГЃЪ§ПЩвдНшжњpHМЦВтЖЈШмвКЕФpHРДШЗЖЈЁЃШєвбжЊШѕЫсЕФНтРыГЃЪ§![]() ЃЌОЭПЩвдМЦЫуГівЛЖЈХЈЖШЕФШѕЫсЕФЦНКтзщГЩЁЃНВНтР§5-1ЁЃ
ЃЌОЭПЩвдМЦЫуГівЛЖЈХЈЖШЕФШѕЫсЕФЦНКтзщГЩЁЃНВНтР§5-1ЁЃ
дкШѕЫсЁЂШѕМюЕФНтРыЦНКтзщГЩМЦЫужаГЃгУЕННтРыЖШ(ІС)ЕФИХФюЃЌЖЈвхШчЯТЃК
![]()
дкЖЈШнЗДгІжаЃЌНтРыЖШЕФБэДяЪНЮЊЃК
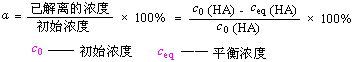
ШѕЫсЕФНтРыЖШЕФДѓаЁвВПЩвдБэЪОЮЊЫсЕФЯрЖдЧПШѕЁЃдкЮТЖШКЭХЈЖШЯрЭЌЕФЧщПіЯТЃЌНтРыЖШДѓЕФЫсЃЌ![]() ДѓЃЌЦфpHаЁЃЌЮЊНЯЧПЫсЃЛНтРыЖШаЁЕФЫсЃЌ
ДѓЃЌЦфpHаЁЃЌЮЊНЯЧПЫсЃЛНтРыЖШаЁЕФЫсЃЌ![]() аЁЃЌЦфpHДѓЃЌЮЊНЯШѕЫсЁЃ
аЁЃЌЦфpHДѓЃЌЮЊНЯШѕЫсЁЃ
ЯЁЪЭЖЈТЩЃКдквЛЖЈЮТЖШЯТЃЈ![]() ЮЊЖЈжЕЃЉЃЌФГШѕЕчНтжЪЕФНтРыЖШЫцзХЦфШмвКЕФЯЁЪЭЖјдіДѓЁЃ
ЮЊЖЈжЕЃЉЃЌФГШѕЕчНтжЪЕФНтРыЖШЫцзХЦфШмвКЕФЯЁЪЭЖјдіДѓЁЃ
ЖрдЊШѕЫсЕФНтРыЃЌЮвУЧЕУГівдЯТНсТлЃК
ЃЈ1ЃЉЖрдЊШѕЫсЕФНтРыЪЧЗжВННјааЕФЃЌвЛАу![]() ШмвКжаЕФH+жївЊРДздгкШѕЫсЕФЕквЛВНЕчРыЃЌМЦЫуc(H3O+)ЛђpHЪБПЩжЛПМТЧЕквЛВНЕчРыЁЃ
ШмвКжаЕФH+жївЊРДздгкШѕЫсЕФЕквЛВНЕчРыЃЌМЦЫуc(H3O+)ЛђpHЪБПЩжЛПМТЧЕквЛВНЕчРыЁЃ
ЃЈ2ЃЉЖдгкЖўдЊШѕЫсЃЌЕБ![]() ЪБЃЌc(ЫсИљРызг)Ёж
ЪБЃЌc(ЫсИљРызг)Ёж![]() ,ЖјгыШѕЫсЕФГѕЪМХЈЖШЮоЙиЁЃ
,ЖјгыШѕЫсЕФГѕЪМХЈЖШЮоЙиЁЃ
ЃЈ3)ЖдгкЖўдЊШѕЫсЃЌШєc(ШѕЫс)вЛЖЈЪБЃЌc(ЫсИљРызг)гыc(H3O+)ГЩе§БШЁЃ
ЯТУцЮвУЧЗжБ№бЇЯАВЛЭЌбЮШмвКЕФЫсМюЦНКтЃК
1.ЧПЫсЧПМюбЮЃЈРызгЫсЃЉЃЌР§ШчЃЌNH4ClдкЫЎжаШЋВПНтРыЃК
![]()
Cl-(aq)ВЛгыЫЎЗДгІЃЌЖјNH4+(aq)гыH2OЗДгІЃЈЫЎНтЗДгІЃЉ
![]()
ИУЗДгІжЪзгзЊвЦЗДгІжаЃЌNH4+ЪЧжЪзгЫсЃЌЦфНтРыГЃЪ§ЮЊЃК
![]()
![]() вВНаNH4+ЕФЫЎНтГЃЪ§ЃЈвВМЧзї
вВНаNH4+ЕФЫЎНтГЃЪ§ЃЈвВМЧзї![]() ЃЉ
ЃЉ
NH4+КЭNH3ЪЧЙВщюЫсМюЖдЁЃЦфЪЕШЮКЮвЛЖдЙВщюЫсМюЕФНтРыГЃЪ§ЖМЗћКЯетвЛЙиЯЕЃЌЭЈЪНЮЊЃК
![]()
СНБпЗжБ№ШЁИКЖдЪ§ЃК
![]()
25ЁцЯТЃЌ![]()
жЪзгЫсNH4+ЕФНтРыЖШОЭЪЧЫљЮНЕФбЮРрЕФЫЎНтЖШЁЃ
![]()
![]()
гЩЩЯЗжЮіПЩжЊЃЌЧПЫсШѕМюбЮЕФЦНКтЮЪЬтЃЌЭъШЋзЊЛЏГЩСЫШѕЫсШмвКЕФЦНКтЮЪЬтЁЃ
2.ШѕЫсЧПМюбЮЃЈРызгМюЃЉЃКШѕЫсШѕМюбЮдкЫЎШмвКжаГЪМюадЃЌЪЧвђЮЊШѕЫсИљРызгдкЫЎжаЗЂЩњЫЎНтЕФдЕЙЪЁЃР§ШчNaAcЃЌШмгкЫЎЭъШЋНтРыЁЃ
![]()
Ac-ЗЂЩњЫЎНтЗДгІЃЈжЪзгзЊвЦЗДгІЃЉ
![]()
![]()
![]() ЁЊЁЊжЪзгМюAc-ЕФНтРыГЃЪ§ЃЌвВНаAc-ЕФЫЎНтГЃЪ§ЃЈвВМЧзї
ЁЊЁЊжЪзгМюAc-ЕФНтРыГЃЪ§ЃЌвВНаAc-ЕФЫЎНтГЃЪ§ЃЈвВМЧзї![]() ЃЉЁЃ
ЃЉЁЃ
ЖрдЊШѕЫсЧПМюбЮЕФвѕРызгдкЫЎжаЕФНтРыЪЧЗжВННјааЕФЁЃШчЃКNa3PO4ЕФЫЎНт
![]()
![]()
![]()
![]()
![]()
![]()
![]() ЃЌЖрдЊШѕЫсЧПМюбЮЮЪЬтОЭзЊЛЏЮЊЖрдЊШѕМюЕФШмвКЦНКтЮЪЬтЁЃ
ЃЌЖрдЊШѕЫсЧПМюбЮЮЪЬтОЭзЊЛЏЮЊЖрдЊШѕМюЕФШмвКЦНКтЮЪЬтЁЃ
ЖрдЊЫсЕФЫсЪНбЮЃЌШчЃКNaHCO3,NaH2PO4,Na2HPO4ЕШЃЌШмгкЫЎКѓЭъШЋНтРыЩњГЩЕФвѕРызгHCO3-,H2PO4-,HPO4-ЕШМШФмИјГіжЪзггжФмНгЪмжЪзгЃЌЪЧСНадЃЌЦфЫЎШмвКМШгаМюадЕФЃЌвВгаЫсадЕФЁЃ
ШчКЮДгРэТлЩЯХаЖЯЦфЫсМюадФиЃПР§ШчЃКСзЫсЖўЧтФЦNaH2PO4ШмвКжаЃЌH2PO4-ЕФНтРыЦНКтЃК
![]()
![]()
H2PO4-ЕФЫЎНтЦНКтЃК
![]()
![]()
НтРыДѓгкЫЎНтЃЌNaH2PO4ШмвКЯдЫсадЃЛЯрЗДЃЌNa2HPO4ШмвКЫЎНтДѓгкНтРыЃЌЯдШѕМюадЁЃ
4.ШѕЫсШѕМюбЮЃКNH4AcШмгкЫЎЭъШЋНтРыЃЌНтРыГіЕФNH4+ЃЌAc-ЭЌЪБЫЎНтЃЌДцдкЯТСаЦНКтЃК
![]()
Шє![]() ШмвКГЪЫсадNH4F
ШмвКГЪЫсадNH4F![]()
Шє![]() ШмвКГЪжаадNH4Ac
ШмвКГЪжаадNH4Ac![]()
Шє![]() ШмвКГЪМюадNH4CN
ШмвКГЪМюадNH4CN![]()
5.гАЯьбЮРрЫЎНтЕФвђЫигабЮЕФХЈЖШЁЂЮТЖШЁЂШмвКЕФЫсМюЖШЕШЁЃ
бЮЕФХЈЖШЃКЯЁЪЭгаРћгкЫЎНтЁЃ
ЮТЖШЃКМгШШгаРћгкЫЎНтЁЃбЮРрЫЎНтЗДгІЪЧжаКЭЗДгІЕФФцЗДгІЃЌвђДЫЪЧЮќШШЗДгІЃЌЫцзХЮТЖШЩ§ИпЃЌ
ЫЎНтГЃЪ§діДѓЃЌЫЎНтМгОчЁЃ
ШмвКЕФЫсМюЖШЃКМгЫсвжжЦЫЎНтЁЃР§ШчЃК
![]()
![]()
![]()
ХфжЦетаЉбЮЕФШмвКЪБЃЌЭЈГЃЯШНЋбЮШмгкНЯХЈЕФЯргІЕФЫсжаЃЌШЛКѓдйМгЫЎЯЁЪЭЕНвЛЖЈХЈЖШЁЃМДРћгУИФБфЫсЖШЕФЗНЗЈЃЌЪЙЫЎНтДяЕНЦНКтЁЃ
дкHAcШмвКжаЃЌДцдкЯТСаЦНКтЃК![]()
ЯђИУШмвКжаМгШыгыHAcКЌгаЯрЭЌРызгЕФЧПЕчНтжЪNH4AcЃЌc(Ac-)діМгЃЌЩЯЦНКтЯђзѓвЦЖЏЃЌМДHAcЕФНтРыЖШНЋМѕаЁЁЃЯёетбљдкШѕЕчНтжЪШмвКжаЃЌМгШыгыЦфКЌгаЯрЭЌРызгЕФвзШмЧПЕчНтжЪЖјЪЙШѕЕчНтжЪЕФНтРыЖШНЕЕЭЕФЯжЯѓГЦЮЊЭЌРызгаЇгІЁЃ
ЧАУцвбОМЦЫуГіЃЌ0.10molЁЄL-1HAcШмвКpH=2.89ЃЌІС=1.3%ЁЃПЩМћЃЌHAcЕФНтРыЖШЕФШЗНЕЕЭСЫКмЖрЁЃ
ЪзЯШЃЌЮвУЧЯШЗжЮіЯТБэЕФЪЕбщЪ§ОнЁЃ
| 1.8ЁС10-5molЁЄL-1HCl | 0.10molЁЄL-1HAc-0.10molЁЄL-1NaAc |
1.0LШмвКЕФpHжЕ | 4.74 | 4.74 |
Мг0.010molNaOH(s)Кѓ | 12.00 | 4.83 |
Мг0.010molHClКѓ | 2.00 | 4.66 |
дкЯЁбЮЫсжа(1.8ЁС10-5molЁЄL-1)ШмвКжаЃЌМгШыЩйСПNaOHЛђHClЃЌpHгаНЯУїЯдЕФБфЛЏЃЌЫЕУїетжжШмвКВЛОпгаБЃГжpHЯрЖдЮШЖЈЕФадФмЁЃЕЋдкетЖдЙВщюЫсМюзщГЩЕФШмвКжаЃЌМгШыЩйСПЕФЧПЫсЛђЧПМюЃЌШмвКЕФpHИФБфКмаЁЁЃетРрШмвКОпгаЛКНтИФБфЧтРызгХЈЖШЖјБЃГжpHЛљБОВЛБфЕФадФмЁЃ
ОпгаФмБЃГжpHЯрЖдЮШЖЈадФмЕФШмМСЃЌМДВЛвђМгШыЩйСПЧПЫсКЭЧПМюЖјЯджјИФБфЕФШмвКНаЛКГхШмвКЁЃ
ЛКГхШмвКжаДцдкЕФжЪзгзЊвЦЗДгІЮЊЃК![]()
![]()
ШмвКжаHAЁЂA-ЪЧДѓСПЕФЃЌH3O+ЪЧЩйСПЕФЃЌc(H3O+)ШЁОігкc(HA)/c(A-)ЁЃ
ЕБМгШыЩйСПЧПМюЃЌЗЂЩњЧПМюгыЧПЫсЕФжаКЭЗДгІЃК![]()
c(A-)ТдгадіМгЃЌc(HA)ТдгаМѕЩйЃЌЕЋвђШмвКжаHAЁЂA-ЪЧДѓСПЕФЃЌc(HA)/c(A-)БфЛЏВЛДѓЃЌвђДЫШмвКжаc(H3O+)ЛђpHЛљБОВЛБфЁЃ
ЕБМгШыЩйСПЧПЫсЃЌНЋЗЂЩњШчЯТжаКЭЗДгІЃК![]()
ЪЙШмвКжаc(A-)ТдгаМѕЩйЃЌc(HA)ТдгадіМгЃЌЕЋвђШмвКжаHAЁЂA-ЪЧДѓСПЕФЃЌc(HA)/c(A-)БфЛЏВЛДѓЃЌвђДЫШмвКжаc(H3O+)ЛђpHЛљБОВЛБфЁЃ
гЩЛКГхШмвКдРэПЩжЊЃК![]()
НЋЕШЪНСНБпЗжБ№ШЁИКЖдЪ§ЃК![]() Лђ
Лђ![]()
ЖдЙВщюЫсМюРДЫЕЃЌ25ЁцЪБЃЌ![]() ЁЂ
ЁЂ![]()
зЂвтЃКc(HA)КЭc(A-)ЪЧЙВщюЫсЁЂМюЕФЦНКтХЈЖШЃЌЕЋгЩгкЭЌРызгаЇгІЕФДцдкЃЌГ§СЫ![]() ЕФЧщПіЭтЃЌЭЈГЃгУГѕЪМХЈЖШc0(HA)КЭc0(A-)ДњжЎЁЃдкИїжжЫсМюШмвКжаЃЌжСЩйгавЛжжЫсЃЈЛђМюЃЉгыШмМСЫЎЕФНтРыЗДгІЭЌЪБДцдкЃЌH+ЛђOHЃЭЌЪБВЮгыСНЃЈЖрЃЉжжНтРыЦНКтЁЃдкМЦЫуpHЪБвЊЩЦгкзЅзЁжїЕМЗДгІЃЌМДвРОнЩњГЩcH+ЛђcOHЃДѓЕФНтРыЦНКтРДМЦЫуЯЕЭГЕФзщГЩЁЃДЫЭтЃЌдкМЦЫуШѕЫс,ШѕМюШмвКЕФpHжЕЪБЃЌжЛгаШмвКХЈЖШВЛЬЋЯЁЃЈШчДѓгк10Ѓ4molЁЄLЃ1ЃЉЪБЃЌВХФмВЛПМТЧЫЎЕФНтРыЦНКтЁЃ
ЕФЧщПіЭтЃЌЭЈГЃгУГѕЪМХЈЖШc0(HA)КЭc0(A-)ДњжЎЁЃдкИїжжЫсМюШмвКжаЃЌжСЩйгавЛжжЫсЃЈЛђМюЃЉгыШмМСЫЎЕФНтРыЗДгІЭЌЪБДцдкЃЌH+ЛђOHЃЭЌЪБВЮгыСНЃЈЖрЃЉжжНтРыЦНКтЁЃдкМЦЫуpHЪБвЊЩЦгкзЅзЁжїЕМЗДгІЃЌМДвРОнЩњГЩcH+ЛђcOHЃДѓЕФНтРыЦНКтРДМЦЫуЯЕЭГЕФзщГЩЁЃДЫЭтЃЌдкМЦЫуШѕЫс,ШѕМюШмвКЕФpHжЕЪБЃЌжЛгаШмвКХЈЖШВЛЬЋЯЁЃЈШчДѓгк10Ѓ4molЁЄLЃ1ЃЉЪБЃЌВХФмВЛПМТЧЫЎЕФНтРыЦНКтЁЃ
ЬсЮЪбЇЩњЫМПМЃКШєгЩNaH2PO4гыNa2HPO4ЙЙГЩЛКГхЯЕЭГЃЌШчКЮМЦЫуШмвКЕФpHжЕЃП
ЬсЪОЃК![]()
ЛКГхШмвКЕФpHжївЊЪЧгЩ![]() Лђ
Лђ![]() ОіЖЈЕФЃЌЛЙгыc(A-)/c(HA)гаЙиЁЃЕБc(A-)/c(HA)=1/10ЁЋ10/1ЪБЃЌЛКГхзїгУгааЇЃЌДЫЗЖЮЇНазіЛКГхЗЖЮЇЁЃЛКГхШмвКЕФЛКГхФмСІЪЧгаЯоЕФЁЃЗжЮіЛЏбЇжаЖЈвхЃКЪЙЛКГхШмвКЕФpHжЕИФБф1.0ЫљашЕФЧПЫсЛђЧПМюЕФСПЃЌГЦЮЊЛКГхФмСІЁЃЕБc(A-)/c(HA)НгНќ1ЪБЃЌЛКГхФмСІДѓЁЃзмжЎЃЌвЊЪЙЛКГхгааЇЃЌВЛНігІЪЙЛКГхШмвКpHдкШмвКЗЖЮЇжЎФкЖјЧвгІОЁПЩФмНгНќ
ОіЖЈЕФЃЌЛЙгыc(A-)/c(HA)гаЙиЁЃЕБc(A-)/c(HA)=1/10ЁЋ10/1ЪБЃЌЛКГхзїгУгааЇЃЌДЫЗЖЮЇНазіЛКГхЗЖЮЇЁЃЛКГхШмвКЕФЛКГхФмСІЪЧгаЯоЕФЁЃЗжЮіЛЏбЇжаЖЈвхЃКЪЙЛКГхШмвКЕФpHжЕИФБф1.0ЫљашЕФЧПЫсЛђЧПМюЕФСПЃЌГЦЮЊЛКГхФмСІЁЃЕБc(A-)/c(HA)НгНќ1ЪБЃЌЛКГхФмСІДѓЁЃзмжЎЃЌвЊЪЙЛКГхгааЇЃЌВЛНігІЪЙЛКГхШмвКpHдкШмвКЗЖЮЇжЎФкЖјЧвгІОЁПЩФмНгНќ![]() ЁЃДЫЭтЃЌЙВщюЫсМюЕФХЈЖШгІЪЪЕБЕФДѓЃЌВХФмБЃжЄНЯЧПЕФЛКГхФмСІЁЃ
ЁЃДЫЭтЃЌЙВщюЫсМюЕФХЈЖШгІЪЪЕБЕФДѓЃЌВХФмБЃжЄНЯЧПЕФЛКГхФмСІЁЃ
ЛКГхШмвКЕФбЁдёХфжЦддђЮЊЃК
ЃЈ1ЃЉЛКГхШмвКГ§СЫВЮгыКЭH+ЛђOH-гаЙиЕФЗДгІЭтЃЌВЛФмгыЗДгІЯЕЭГжаЕФЦфЫћЮяжЪЗЂЩњИБЗДгІЁЃ
ЃЈ2ЃЉ![]() Лђ
Лђ![]() ОЁПЩФмНгНќЫљашШмвКЕФpHЁЃ
ОЁПЩФмНгНќЫљашШмвКЕФpHЁЃ
аЁНсЃКЧыЭЌбЇУЧжиЕуеЦЮевЛдЊШѕЫсЃЈМюЃЉЕФНтРыЦНКтМАЦфЦНКтзщГЩЕФМЦЫуЃЌЛКГхШмвКpHжЕЕФМЦЫуЁЃЧыЭЈЙ§ПЮКѓЯАЬтЕФСЗЯАМгвдЙЎЙЬЫљбЇжЊЪЖЕуЁЃ
дкЩЯвЛНкжаЃЌЮвУЧЯЕЭГбЇЯАСЫЫЎШмвКжаЕФЫсМюЦНКтЃЌетЪЧвЛжжОљЯрЗДгІЁЃГ§ДЫжЎЭтЃЌСэвЛРрживЊЕФРызгЗДгІЪЧФбШмЕчНтжЪдкЫЎжаЕФШмНтЃЌМДдкКЌгаЙЬЬхФбШмЕчНтжЪЕФБЅКЭШмвКжаЃЌДцдкзХЕчНтжЪгыгЩЫќНтРыВњЩњЕФРызгжЎМфЕФЦНКтЃЌНазіГСЕэШмНтЦНКтЁЃетЪЧвЛжжЖрЯрРызгЦНКтЁЃГСЕэЕФЩњГЩКЭШмНтЯжЯѓдкЮвУЧЕФжмЮЇОГЃЗЂЩњЁЃР§ШчШЫЬхФкЩіНсЪЏЕФаЮГЩЃЌОЭЪЧФбШмбЮВнЫсИЦКЭСзЫсИЦЕФЩњГЩЃЛздШЛНчжаЕФжгШщЪЏЕФаЮГЩЃЌОЭЪЧЬМЫсИЦГСЕэЕФЩњГЩКЭШмНтЗДгІгаЙиЃЛЙЄвЕЩЯЃЌПЩвдгУЬМЫсФЦКЭЯћЪЏЛвжЦШЁЩеМюЕШЁЃетаЉЪЕР§ЫЕУїЃЌГСЕэ-ШмНтЦНКтЖдЩњЮяЛЏбЇЁЂвНбЇЁЂЙЄвЕЩњВњЕШгазХЩюдЖгАЯьЃЌПЩвдЬсЙЉРэТлжИЕМЁЃ
ШмНтадЪЧЮяжЪживЊЕФаджЪжЎвЛЃЌГЃвдШмНтЖШРДЖЈСПБъУїЮяжЪЕФШмНтЖШЁЃдквЛЖЈЮТЖШЯТЃЌДяЕНШмНтЦНКтЪБЃЌвЛЖЈЕФШмМСжаКЌгаШмжЪЕФжЪСПЃЌНазїШмНтЖШЃЌЭЈГЃвдЗћКХSБэЪОЁЃЖдгкЫЎШмвКРДЫЕЃЌЭЈГЃвдБЅКЭШмвКжаУП100gЫЎЫљКЌШмжЪЕФжЪСПРДБэЪОЃЌМДЃКвд"g/100gЫЎ"БэЪОЁЃаэЖрЮоЛњЛЏКЯЮядкЫЎжаШмНтЪБЃЌФмаЮГЩЫЎКЯбєРызгКЭвѕРызгЃЌГЦЦфЮЊЕчНтжЪЃЌЕчНтжЪЕФШмНтЖШгаКмДѓЕФВювьЃЌвЛАуЗжЮЊПЩШмЁЂЮЂШмКЭФбШмЕШВЛШнЕФЕШМЖЁЃ
дквЛЖЈЮТЖШЯТЃЌНЋФбШмЕчНтжЪОЇЬхЗХШыЫЎжаЃЌОЭЗЂЩњШмНтКЭГСЕэСНИіЙ§ГЬЁЃЕБШмНтКЭГСЕэЫйТЪЯрЕШЪБ,БуНЈСЂСЫвЛжжЖЏЬЌЕФЖрЯрЦНКтЁЃвдBaSO4ОЇЬхШмгкЫЎаЮГЩЫЎКЯРызгЕФЙ§ГЬЮЊР§,ПЩБэЪОЮЊЃК
![]()
ЦНКтГЃЪ§ЮЊЃК![]()
![]() НазіШмЖШЛ§ГЃЪ§,МђГЦШмЖШЛ§ЁЃ
НазіШмЖШЛ§ГЃЪ§,МђГЦШмЖШЛ§ЁЃ
ЖдгквЛАуЕФГСЕэЗДгІЃК![]()
ШмЖШЛ§ГЃЪ§ЕФЭЈЪНЃК![]()
МДЃЌШмЖШЛ§ЕШгкГСЕэ-ШмНтЦНКтЪБРызгХЈЖШУнЕФГЫЛ§ЃЌУПжжРызгХЈЖШЕФУнгыЛЏбЇМЦСПЪНжаЕФМЦСПЪ§ЯрЕШЁЃвЊЬиБ№зЂвтЃКKspӨ�������ЕФЪ§жЕдкЯЁШмвКжаВЛЪмЦфЫћРызгЕФДцдкЕФгАЯь,жЛШЁОігкЮТЖШЁЃЮТЖШЩ§ИпЃЌЖрЪ§ФбШмЛЏКЯЮяЕФШмЖШЛ§діДѓЁЃ
ШмЖШЛ§КЭШмНтЖШЖМПЩвдгУРДБэЪОФбШмЕчНтжЪЕФШмНтадЁЃСНепМШгаСЊЯЕЃЌгжгаЧјБ№ЁЃдкЯрЙиШмЖШЛ§ЕФМЦЫужаЃЌРызгХЈЖШБиаыЪЧЮяжЪЕФСПХЈЖШЃЌЦфЕЅЮЛЪЧmolЁЄL-1ЃЌЖјШмНтЖШЕФЕЅЮЛЭљЭљЪЧg/100gЫЎЁЃвђДЫЃЌМЦЫуЪБгаЪБвЊЯШНЋФбШмЕчНтжЪЕФШмНтЖШSЕФЕЅЮЛЛЛЫуmolЁЄL-1ЁЃ
![]()
ЦНКтХЈЖШ/molЁЄL-1 nS mS
![]()
ЖдгкABаЭЃК![]()
ЖдгкAB2аЭЃК![]()
ФбШмЕчНтжЪЕФГСЕэ-ШмНтЦНКтгыЦфЫћЖЏЬЌЦНКтвЛбљЃЌЭъШЋзёбLe ChatelierдРэЁЃЖдгкФбШмЕчНтжЪЕФЖрЯрРызгЦНКтЮяжЪЃКЗДгІЩЬJ={c(Am+)}n{c(Bn-)}mГСЕэ-ШмНтЦНКтЕФЗДгІЩЬХаОнЃЌЃЈЗДгІЩЬгжБЛГЦЮЊФбШмЕчНтжЪЕФРызгЛ§ЃЉМДШмЖШЛ§ЙцдђЃК
![]() ЃЌЦНКтЯђзѓвЦЖЏЃЌГСЕэЮіГіЃЛ
ЃЌЦНКтЯђзѓвЦЖЏЃЌГСЕэЮіГіЃЛ
![]() ЃЌДІгкЦНКтзДЬЌЃЌБЅКЭШмвКЃЛ
ЃЌДІгкЦНКтзДЬЌЃЌБЅКЭШмвКЃЛ
![]() ЃЌЦНКтЯђгввЦЖЏЃЌЮоГСЕэЮіГіЃЌШєдРДгаГСЕэДцдкЃЌдђГСЕэШмНтЁЃ
ЃЌЦНКтЯђгввЦЖЏЃЌЮоГСЕэЮіГіЃЌШєдРДгаГСЕэДцдкЃЌдђГСЕэШмНтЁЃ
дкФбШмЕчНтжЪБЅКЭШмвКжаЃЌМгШыКЌгаЯрЭЌРызгЕФвзШмЧПЕчНтжЪЃЌЖјЪЙФбШмЕчНтжЪЕФШмНтЖШНЕЕЭЕФзїгУГЦЮЊЭЌРызгаЇгІЁЃетгыЩЯвЛеТдкЛКГхвКФЧвЛНкЫљбЇЕФШѕЫсЛђШѕМюЕФЭЌРызгаЇгІвВЪЧвЛжБЕФЁЃ
дкЪЕбщжа,ГЃГЃРћгУМгШыЪЪЕБЙ§СПЕФГСЕэМС,ЪЙГСЕэЧїгкЭъШЋЁЃЕЋЪЧзЂвтЃЌШчЙћМгШыГСЕэМСЬЋЖрЪБЃЌВЛНіВЛЛсВњЩњУїЯдЕФЭЌРызгаЇгІЃЌЭљЭљЛЙЛсВњЩњЯрЗДЕФзїгУ,ЪЙГСЕэЕФШмНтЖШдіДѓЃЌетжжгАЯьжЎвЛОЭЪЧбЮаЇгІЁЃ
ЙлВьБэ6-2ЃЌAgClдкKNO3ШмвКжаЕФШмНтЖШ(25Ёц)ЁЃПЩМћЃЌAgClдкKNO3ШмвКжаЕФШмНтЖШБШЦфЫќДПЫЎжаЕФШмНтЖШДѓЃЌВЂЧвKNO3ЕФХЈЖШдНДѓЃЌAgClШмНтЖШвВдНДѓЁЃ етжжгЩгкМгШывзШмЧПЕчНтжЪЖјЪЙФбШмЕчНтжЪШмНтЖШдіДѓЕФаЇгІ,НазібЮаЇгІЁЃ зЂвтЃКМгШыОпгаЯрЭЌРызгЕФЕчНтжЪЃЌдкВњЩњЭЌРызгаЇгІЕФЭЌЪБЃЌвВФмВњЩњбЮаЇгІЁЃ
ЗжЮіБэ6-3ЃЌPbSO4дкNa2SO4ШмвКжаЕФШмНтЖШЃКЕБNa2SO4ЕФХЈЖШДг0діМгЕН0.04molЁЄL-1ЪБЃЌPbSO4ШмНтЖШж№НЅБфаЁЃЌЭЌРызгаЇгІЦ№жїЕМзїгУЃЌЕБNa2SO4ЕФХЈЖШДѓЮЊ0.04molЁЄL-1ЪБЃЌPbSO4ЕФШмНтЖШзюаЁЃЛЕБNa2SO4ЕФХЈЖШДѓгк0.04molЁЄL-1ЪБЃЌPbSO4ЕФШмНтЖШж№НЅдіДѓЃЌбЮаЇгІЦ№жїЕМзїгУЁЃвЛАуЫЕРДЃЌШєФбШмЕчНтжЪЕФШмЖШЛ§КмаЁЪБЃЌбЮаЇгІЕФгАЯьКмаЁЃЌПЩКіТдВЛМЦЃЛШєФбШмЕчНтжЪЕФШмЖШЛ§НЯДѓЪБ,ШмвКжаИїжжРызгЕФзмХЈЖШвВНЯДѓЪБЃЌОЭгІИУПМТЧбЮаЇгІЕФгАЯьЁЃ
ШчЙћФбШмЕчНтжЪMAЕФвѕРызгЪЧФГШѕЫсЕФЙВщюМюЃЌгЩгкЙВщюМюЖджЪзггаНЯЧПЕФЧзКЭФмСІЃЌдђЫќУЧЕФШмНтЖШНЋЫцpHЕФМѕаЁЖјдіДѓЁЃетРрФбШмЕчНтжЪОЭЪЧЭЈГЃЫљЫЕЕФФбШмШѕЫсбЮКЭФбШмН№ЪєЧтбѕЛЏЮяЁЃЧтбѕИљРызгЪЧЫЎжаЕФзюЧПМюЃЌЫќЪЧШѕЫсЫЎЕФЙВщюМюЃЌДгетИівтвхЩЯЫЕЃЌН№ЪєЧтбѕЛЏЮявВЪЧШѕЫсбЮЁЃРћгУШѕЫсбЮдкЫсжаШмНтЖШЕФВювьЃЌЮвУЧПЩвдЭЈЙ§ПижЦШмвКЕФpHРДДяЕНЗжРыН№ЪєРызгЕФФПЕФЁЃ
1.ФбШмН№ЪєЧтбѕЛЏЮяШмгкЫсЃЌГСЕэШмНтЦНКтЃК
![]()
![]()
РћгУЩЯЪНПЩвдМЦЫуЧтбѕЛЏЮяПЊЪМГСЕэКЭГСЕэЭъШЋЪБШмвКЕФc(OH-)ЃЌДгЖјЧѓГіЯргІЬѕМўЕФpHжЕЁЃ
ПЊЪМГСЕэЪБЃК
![]()
c0(Mn+)----ШмвКжаMn+РызгЕФЦ№ЪМХЈЖШ
МДШмвКжаЧтбѕИљРызгЕФХЈЖШаЁгкcЪМ(OH-)ЃЌОЭВЛаЮГЩMn(OH)nГСЕэЃЛШєШмвКжагаГСЕэЃЌжЛвЊНЋШмвКЕФЧтбѕИљРызгПижЦдкcЪМ(OH-)вдЯТЃЌдгаЕФM(OH)nГСЕэНЋШмНтЃЌЧвШмНтКѓШмвКжаMn+РызгХЈЖШЮЊc0(Mn+)ЁЃ
ГСЕэЭъШЋЪБЃК
![]()
ЗжЮіЛЏбЇжаШЯЮЊЃЌЕБШмвКжаРызгХЈЖШаЁгк1.0ЁС10-5molЁЄL-1ЪБЃЌШЯЮЊетжжРызгвбГСЕэЭъШЋЁЃРћгУВЛЭЌРызгаЮГЩЧтбѕЛЏЮяГСЕэКЭГСЕэЭъШЋЪБШмвКЕФpHжЕЕФВювьЃЌПЩНЋВЛЭЌЕФРызгНјааЗжРыЁЃ
вЛаЉСНадЧтбѕЛЏЮяЃЌМШПЩвдШмгкЫсЃЌгжПЩвдШмгкМюЃЌФЧШмвКpHжЕдквЛЖЈЧјМфФкЃЌЛЏКЯЮяЪЧвдЧтбѕЛЏЮяГСЕэДцдкЕФЁЃР§ШчЃК
![]()
![]()
pHдк4ЁЋ11ЗЖЮЇФкAl(OH)3ЛљБОВЛШмНтЁЃЖдгкKspӨ�������ВЛЪЧКмаЁЃЈKspӨ�������ЃК10-12ЁЋ10-13ЃЉЕФФбШмН№ЪєЧтбѕЛЏЮяЃЌГЃЪЙгУАБ-яЇбЮЛКГхШмвКРДПижЦpHжЕЃЌДяЕНГСЕэЕФЩњГЩЛђШмНтЕФФПЕФЁЃ
Н№ЪєСђЛЏЮяЃККмЖрН№ЪєСђЛЏЮядкЫЎжаЖМЪЧФбШмЕФЃЌЖјЧвЫќУЧЕФШмЖШЛ§ГЃЪ§БЫДЫгавЛЖЈЕФВювьЃЌВЂИїгаЬиЖЈЕФбеЩЋЁЃдкЪЕМЪгІгУжаЃЌГЃРћгУСђЛЏЮяШмЖШЛ§ЕФВювьвдМАСђЛЏЮяЕФЬиеїбеЩЋ,РДЗжРыЛђМјЖЈФГаЉН№ЪєРызгЁЃзюНќбаОПБэУї,S2-ЯёO2-вЛбљЪЧКмЧПЕФШѕМю,дкЫЎжаВЛФмДцдкЁЃЮіГіФбШмН№ЪєСђЛЏЮяMSЕФЖрЯрРызгЦНКтВЛФмаДзїЃК
MS(s)![]() M2+(aq)+S2-(aq)
M2+(aq)+S2-(aq)
БиаыПМТЧЧПМюS2-ЖджЪзгЕФЧзКЭзїгУЃЌ
S2-(aq)+H2O(l)![]() HS-(aq)+OH-(aq)
HS-(aq)+OH-(aq)
ЫљвдЃЌФбШмН№ЪєСђЛЏЮяЕФЖрЯрРызгЦНКтгІаДзїЃК
MS(s)+H2O(l)![]() M2+(aq)+OH-(aq)+HS-(aq)
M2+(aq)+OH-(aq)+HS-(aq)
Ky={c(M2+)}{c(OH-)}{c(HS-)}
ЖјФбШмН№ЪєСђЛЏЮядкЫсжаЕФГСЕэ-ШмНтЦНКтЮЊЃК
MS(s)+2H+(aq)![]() M2+(aq)+H2S(aq)
M2+(aq)+H2S(aq)
![]()
Лђ![]()
![]() -ФбШмН№ЪєСђЛЏЮядкЫсжаЕФШмЖШЛ§ГЃЪ§ЁЃ
-ФбШмН№ЪєСђЛЏЮядкЫсжаЕФШмЖШЛ§ГЃЪ§ЁЃ
аэЖрФбШмЛЏКЯЮядкХфЮЛМСЕФзїгУЯТЃЌФмЙЛЩњГЩХфРызгЖјШмНтЁЃР§ШчЃК
![]()
![]()
![]()
вЛАуЧщПіЯТЃЌЕБФбШмЛЏКЯЮяЕФШмЖШЛ§ВЛКмаЁЃЌВЂЧвХфКЯЮяЕФЩњГЩГЃЪ§БШНЯДѓЪБЃЌОЭгаРћгкХфЮЛШмНтЗДгІЕФЗЂЩњЁЃДЫЭтЃЌХфЮЛМСЕФХЈЖШвВЪЧгАЯьФбШмЛЏКЯЮяФмЗёЗЂЩњХфЮЛШмНтЕФживЊвђЫижЎвЛЁЃ
ЧАУцЮвУЧЬжТлЕФЖМЪЧМгвЛжжЪдМСжЛФмЪЙвЛжжРызгЩњГЩГСЕэЕФЧщПіЁЃЪЕМЪЩЯЃЌШмвКжаЭљЭљКЌгаЖржжПЩБЛГСЕэЕФРызгЃЌМДЕБМгШыФГжжГСЕэЪдМСЕФЪБКђЃЌПЩФмЗжБ№КЭШмвКжаЖржжРызгЗЂЩњЗДгІЖјВњЩњГСЕэЁЃдкетжжЧщПіЯТЃЌЕБШмвКжаДцдкЖржжПЩБЛГСЕэЕФРызгЪБЃЌМгШыГСЕэМСЛсВњЩњдѕбљЕФНсЙћФиЃПЪЧЭЌЪБГСЕэЯТРДФиЃПЛЙЪЧвЛВНвЛВНЕиАДЯШКѓДЮађГСЕэФиЃП
дкNa2SКЭK2CrO4ЕШБШР§ЛьКЯЕФШмвКжаЃЌМгШыPb(NO3)2ШмвКЃЌЯШЩњГЩКкЩЋЕФPbSГСЕэЃЌКѓЩњГЩЛЦЩЋЕФPbCrO4ГСЕэЁЃСэЭтЃЌдкКЌгаЯрЭЌХЈЖШЃЈ1ЁС10-3molЁЄL-1)ЕФAg+КЭPb2+ЕФЛьКЯШмвКжаЃЌЯШМгвЛЕЮ0.1molЁЄL-1ЕФK2CrO4ШмвКЃЌДЫЪБжЛгаЛЦЩЋЕФPbCrO4ГСЕэЮіГіЃЛШчЙћМЬајЕЮМгШмвК,ВХгазЉКьЩЋЕФAg2CrO4ГСЕэЮіГіЁЃетжжЯШКѓГСЕэЕФЯжЯѓЃЌНазіЗжВНГСЕэЛђЗжМЖГСЕэЁЃеЦЮеСЫЗжВНГСЕэЕФЙцТЩЃЌОЭПЩИљОнОпЬхЧщПіЃЌЪЪЕБЕФПижЦЬѕМўЃЌДяЕНЗжРыРызгЕФФПЕФЁЃ
гааЉГСЕэМШВЛШмгкЫЎвВВЛШмгкЫсЃЌЛЙЮоЗЈгУХфЮЛШмНтКЭбѕЛЏЛЙдШмНтЕФЗНЗЈАбЫќжБНгШмНтЁЃетЪБЃЌПЩАбвЛжжФбШмЕчНтжЪзЊЛЏЮЊСэвЛжжФбШмЕчНтжЪЃЌШЛКѓЪЙЦфШмНтЁЃетжжАбвЛжжГСЕэзЊЛЏЮЊСэвЛжжГСЕэЕФЙ§ГЬЃЌНазіГСЕэзЊЛЏЁЃШчдкAgNO3КЭK2CrO4ЕФЛьКЯШмвКжаЃЌдйж№ЕЮМгШыNaClШмвКЃЌБпМгБпеёЕДЃЌГСЕэгЩКьБфАзЁЃ
аЁНсЃЌдкБОеТжаЃЌЮвУЧМЬајбЇЯАСЫЙигкЦНКтЕФЬжТлЃЌИУФкШнгыЫсМюЦНКтгаКмЖрЯрЫЦжЎДІЃЌЮвУЧдкПЮЬУЩЯОЭНВЕФУЛФЧУДЯъЯИЃЌЧыЭЌбЇУЧПЮКѓАбЦНКтЯрЙиФкШнСЊЯЕЦ№РДЃЌЫМПМЦфЯыЭЈжЎДІЃЌЖјВЛЪЧЗжПЊЫРМЧгВБГЁЃ
ЕкЫФеТ бѕЛЏЛЙдЗДгІгыгІгУЕчЛЏбЇ
ЮоЛњЛЏбЇЗДгІвЛАуЗжЮЊСНДѓРрЃЌвЛРрЪЧдкЗДгІЙ§ГЬжаЃЌЗДгІЮяжЎМфУЛгаЕчзгЕФзЊвЦЛђЕУЪЇЃЌШчЫсМюЗДгІЁЂГСЕэЗДгІЃЌЫќУЧжЛЪЧРызгЛђдзгМфЕФЯрЛЅНЛЛЛЃЈетвВе§ЪЧЮвУЧЧАУцЫљбЇЕФжЊЪЖЃЉЃЛСэвЛРрдђЪЧдкЗДгІЙ§ГЬжаЃЌЗДгІЮяжЎМфЗЂЩњСЫЕчзгЕФЕУЪЇЛђзЊвЦЃЌетРрЗДгІБЛГЦжЎЮЊбѕЛЏЛЙдЗДгІЁЃбѕЛЏЛЙдЗДгІЕФЪЕжЪЪЧЕчзгЕФЕУЪЇКЭзЊвЦЃЌдЊЫибѕЛЏжЕЕФБфЛЏЪЧЕчзгЕУЪЇЕФНсЙћЁЃдЊЫибѕЛЏжЕЕФИФБфвВЪЧЖЈвхбѕЛЏМСЁЂЛЙдМСКЭХфЦНбѕЛЏЛЙдЗДгІЗНГЬЪНЕФвРОнЁЃ
1970ФъЙњМЪДПЛЏбЇКЭгІгУЛЏбЇбЇЛсЃЈIUPACЃЉЖЈвхбѕЛЏжЕЃЈoxidation numberЃЉЕФИХФюЮЊЃКбѕЛЏжЕЃЈгжГЦбѕЛЏжЕЃЉЪЧФГдЊЫивЛИідзгЕФКЩЕчЪ§ЃЌетжжКЩЕчЪ§ЪЧНЋГЩМќЕчзгжИЖЈИјЕчИКадНЯДѓЕФдзгЖјЧѓЕУЁЃШЗЖЈдЊЫидзгбѕЛЏжЕгаЯТСаддђЃК
ЃЈ1ЃЉЕЅжЪЕФбѕЛЏжЕЮЊСуЁЃвђЭЌвЛдЊЫиЕФЕчИКадЯрЭЌЃЌдкаЮГЩЛЏбЇМќЪБВЛЗЂЩњЕчзгЕФзЊвЦЛђЦЋРыЁЃР§ШчS8жаЕФSЃЌCl2жаЕФClЃЌH2жаЕФHЃЌН№ЪєCuЁЂAlЕШЃЌбѕЛЏжЕОљЮЊСуЁЃ
ЃЈ2ЃЉЧтдкЛЏКЯЮяжаЕФбѕЛЏжЕвЛАуЮЊ1ЃЌЕЋдкЛюЦУН№ЪєЕФЧтЛЏЮяжаЃЌЧтЕФбѕЛЏжЕЮЊЃ1ЃЌШчNaHЃ1ЁЃ
ЃЈ3ЃЉбѕдкЛЏКЯЮяжаЕФбѕЛЏжЕвЛАуЮЊЃ2ЃЌЕЋдкЙ§бѕЛЏЮяжаЃЌбѕЕФбѕЛЏжЕЮЊЃ1ЃЌШчH2OЃ12ЁЂBaOЃ12ЃЛдкГЌбѕЛЏЮяжаЃЌбѕЕФбѕЛЏжЕЮЊЃ1/2ЃЌШчKOЃ1/22ЃЛдкЗњЕФбѕЛЏЮяжаЃЌбѕЕФбѕЛЏжЕЮЊЂђЃЌШчO2F2ЁЃ
ЃЈ4ЃЉЕЅдзгРызгдЊЫиЕФбѕЛЏжЕЕШгкЫќЫљДјЕФЕчКЩЪ§ЁЃШчМюН№ЪєЕФбѕЛЏжЕЮЊ1ЃЌМюЭСН№ЪєЕФбѕЛЏжЕЮЊ2ЁЃ
ЃЈ5ЃЉдкЖрдзгЕФЗжзгжаЫљгадЊЫиЕФдзгбѕЛЏжЕЕФДњЪ§КЭЕШгкСуЃЛдкЖрдзгЕФРызгжаЫљгадЊЫиЕФдзгбѕЛЏжЕЕФДњЪ§КЭЕШгкРызгЫљДјЕФЕчКЩЪ§ЁЃ
ИљОнвдЩЯЙцдђЃЌЮвУЧМШПЩвдМЦЫуЛЏКЯЮяЗжзгжаИїжжзщГЩдЊЫидзгЕФбѕЛЏжЕЃЌврПЩвдМЦЫуЖрдзгРызгжаИїзщГЩдЊЫидзгЕФбѕЛЏжЕЁЃР§ШчЃК
MnO4-жаMnЕФбѕЛЏжЕЮЊЃКx+4ЁС(Ѓ2ЃЉ=Ѓ1x=7
Cr2O72-жаCrЕФбѕЛЏжЕЮЊЃК2x+7ЁС(Ѓ2ЃЉ=Ѓ2x=6
гЩгкбѕЛЏжЕЪЧдкжИЖЈЬѕМўЯТЕФМЦЫуНсЙћЃЌЫљвдбѕЛЏжЕВЛвЛЖЈЪЧећЪ§ЁЃШчдкСЌЫФСђЫсИљРызгЃЈS4O62-ЃЉжаЃЌSЕФбѕЛЏжЕЮЊ5/2ЁЃетЪЧгЩгкЗжзгжаЭЌвЛдЊЫиЕФСђдзгДІгкВЛЭЌЕФбѕЛЏЬЌЃЌЖјАДЩЯЗЈМЦЫуЕФЪЧSдЊЫибѕЛЏжЕЕФЦНОљжЕЃЌЫљвдбѕЛЏжЕгаЗЧећЪ§ГіЯжЁЃ
ИљОнбѕЛЏжЕЕФИХФюЃЌЗВЪЧЮяжЪбѕЛЏжЕЗЂЩњБфЛЏЕФЗДгІЃЌГЦЮЊбѕЛЏЛЙдЗДгІЁЃбѕЛЏжЕЩ§ИпЕФЙ§ГЬГЦЮЊбѕЛЏЃЌбѕЛЏжЕНЕЕЭЕФЙ§ГЬГЦЮЊЛЙдЁЃдкЗДгІЙ§ГЬжаЃЌбѕЛЏжЕЩ§ИпЕФЮяжЪГЦЮЊЛЙдМСЃЈreductantЃЉЃЌбѕЛЏжЕНЕЕЭЕФЮяжЪГЦЮЊбѕЛЏМСЃЈoxidantЃЉЁЃбѕЛЏМСЦ№бѕЛЏзїгУЃЌЫќбѕЛЏЛЙдМСЃЌздЩэБЛЛЙдЃЛЛЙдМСЦ№ЛЙдзїгУЃЌЫќЛЙдбѕЛЏМСЃЌздЩэБЛбѕЛЏЁЃ
бѕЛЏЙ§ГЬЃКбѕЛЏЬЌЩ§ИпЕФЙ§ГЬЃЌЛЙдМС
ЛЙдЙ§ГЬЃКбѕЛЏЬЌНЕЕЭЕФЙ§ГЬЃЌбѕЛЏМС
бѕЛЏаЭЃКИпбѕЛЏЬЌбѕЛЏМС
ЛЙдаЭЃКЕЭбѕЛЏЬЌЛЙдМС
жаМфЬЌЃКМШПЩзїЮЊбѕЛЏМС,гжПЩзіЮЊЛЙдМС
дкбѕЛЏЛЙдЗДгІжаЃЌШєбѕЛЏжЕЕФЩ§ИпКЭНЕЕЭЖМЗЂЩњдкЭЌвЛжжЛЏКЯЮяжаЃЌМДбѕЛЏМСКЭЛЙдМСЮЊЭЌвЛжжЮяжЪЃЌГЦздЩэбѕЛЏЛЙдЗДгІЁЃздЩэбѕЛЏЛЙдЗДгІгжГЦЮЊЦчЛЏЗДгІ(disproportionation reaction)ЁЃ
УПИібѕЛЏЛЙдЗДгІЗНГЬЪНПЩвдВ№ГЩСНИіАыЗДгІЪНЃЌМДЪЇЕчзгЕФбѕЛЏАыЗДгІЪНКЭЕУЕчзгЕФЛЙдАыЗДгІЪНЁЃР§Шч
бѕЛЏЛЙдРызгЗДгІЪНЃКCe4++Fe2+ЃНCe3++Fe3+
бѕЛЏАыЗДгІЪНЃКFe2+ЃeЃНFe3+
ЛЙдАыЗДгІЪНЃКCe4+ЃЋeЃНCe3+
бѕЛЏаЭгыЛЙдаЭЙЙГЩСЫШчЯТСНЖдбѕЛЏЛЙдЕчЖдЃЈredoxcoupleЃЉЃКCe4+/Ce3+КЭFe3+/Fe2+ЁЃвђДЫЃЌбѕЛЏЛЙдЗДгІЪЧСНИіЃЈЛђСНИівдЩЯЃЉбѕЛЏЛЙдЕчЖдЙВЭЌзїгУЕФНсЙћЁЃ
АыЗДгІЪНПЩгУЭЈЪНБэЪОЃКбѕЛЏаЭЃЋeЃНЛЙдаЭ
бѕЛЏЛЙдЕчЖдЪщаДЪБЃЌбѕЛЏаЭаДдкаБЯпзѓВрЃЌЛЙдаЭаДдкаБЯпгвВрЁЃ
бѕЛЏЛЙдЗДгІЗНГЬЪНЕФХфЦНддђЃКЛЙдМСбѕЛЏжЕЩ§ИпЪ§КЭбѕЛЏМСбѕЛЏжЕНЕЕЭЪ§ЯрЕШ(ЕУЪЇЕчзгЪ§ФПЯрЕШЃЉаДГіЛЏбЇЗДгІЗНГЬЪНЃЌШЗЖЈгаЙидЊЫибѕЛЏЬЌЩ§ИпМАНЕЕЭЕФЪ§жЕЃЌШЗЖЈбѕЛЏжЕЩ§ИпМАНЕЕЭЕФЪ§жЕЕФзюаЁЙЋБЖЪ§ЁЃевГібѕЛЏМСЁЂЛЙдМСЕФЯЕЪ§ЃЌКЫЖдЃЌПЩгУH+,OHЈC,H2OХфЦНЁЃ
ЯТУцЗжБ№бЇЯАСНжжХфЦНЕФЗНЗЈЃК1. РызгЕчгкЗЈХфЦНЗНГЬЪНЕФЛљБОВНжшЮЊЃК
(1)аДГіЗДгІЙ§ГЬжабѕЛЏжЕЦ№БфЛЏЕФРызгаДГЩвЛИіУЛгаХфЦНЕФРызгЗНГЬЪНЃК
MnO42-ЃЋSO32-ЁњMn2+ЃЋSO42-
ЃЈ2ЃЉНЋЩЯУцЮДХфЦНЕФРызгЗНГЬЪНЗжаДЮЊСНИіАыЗДгІЪНЃЌвЛИіДњБэбѕЛЏМСЕФЛЙдЗДгІЃЛСэвЛИіДњБэЛЙдМСЕФбѕЛЏЗДгІЃК
MnO42-ЁњMn2+SO32-ЁњSO42-
ЃЈ3ЃЉЗжБ№ХфЦНСНИіАыЗДгІЪНЁЃХфЦНЪБЪзЯШХфЦНдзгЪ§ЃЌШЛКѓдкАыЗДгІЕФзѓБпЛђгвБпМгЩЯЪЪЕБЕчзгЪ§РДХфЦНЕчКЩЪ§ЁЃвдЪЙАыЗДгІЪНСНВрИїжждзгЕФзмЪ§МАОЛЕчКЩЪ§ЯрЕШЯрЕШЁЃ
MnO42-ЛЙдЮЊMn2+ЪБЃЌвЊМѕЩй4ИібѕдзгЃЌдкЫсадНщжЪжаЃЌвЊгы8ИіH+РыгкНсКЯЩњГЩ4ИіH2OЗжзгЁЃ
MnO42-ЃЋ8H+ЁњMn2+ЃЋ4H2O
ЩЯЪНжазѓБпЕФОЛЕчКЩЪ§ЮЊЃЋ7ЃЌгвБпЕФОЛЕчКЩЪ§ЮЊЃЋ2ЃЌЫљвдашдкзѓБпМг5ИіЕчзгЃЌЪЙСНБпЕФЕчКЩЪ§ЯрЕШЃК
MnO42-ЃЋ8H+ЃЋ5e=Mn2+ЃЋ4H2O
SO32-бѕЛЏЮЊSO42-ЪБЃЌдіМгЕФ1ИібѕдзгПЩгЩШмвКжаЕФH2OЗжзгЬсЙЉЃЌЭЌЪБЩњГЩ2ИіH+РызгЃК
SO32-ЃЋH2O=SO42-ЃЋ2H+
ЩЯЪНжаЃЌзѓБпЕФОЛЕчКЩЪ§ЮЊЃ2ЃЌгвБпЕФОЛЕчКЩЪ§ЮЊ0ЃЌЫљвдгвБпгІМгЩЯ2ИіЕчзгЃК
SO32-ЃЋH2O=SO42-ЃЋ2H+ЃЋ2e
ЃЈ4ЃЉИљОнбѕЛЏМСКЭЛЙдМСЕУЪЇЕчзгЪ§БиаыЯрЕШЕФддђЃЌдкСНИіАыЗДгІЪНжаГЫЩЯЯргІЕФЯЕЪ§ЃЈгЩЕУЪЇЕчзгЕФзюаЁЙЋБЖЪ§ШЗЖЈЃЉЃЌШЛКѓСНЪНЯрМгЕУЕНХфЦНЕФРызгЗДгІЗНГЬЪНЁЃ
2MnO42-ЃЋ6H+ЃЋ5SO32-=2Mn2+ЃЋ5SO42-ЃЋ3H2O
бѕЛЏжЕЗЈКЭРыгкЕчзгЗЈИїгагХШБЕуЁЃбѕЛЏжЕЗЈФмНЯбИЫйЕиХфЦНМђЕЅЕФбѕЛЏЛЙдЗДгІЁЃЫќЕФЪЪгУЗЖЮЇНЯЙуЃЌВЛжЛЯогкЫЎШмвКжаЕФЗДгІЃЌЬиБ№ЖдИпЮТЗДгІМАШлШкЬЌЮяжЪМфЕФЗДгІИќЮЊЪЪгУЁЃЖјРызгЕчгкЗЈФмЗДгГГіЫЎШмвКжаЗДгІЕФЪЕжЪЃЌЬиБ№ЖдгаНщжЪВЮМгЕФИДдгЗДгІХфЦНБШНЯЗНБуЁЃЕЋЪЧЃЌРызгЕчзгЗЈНіЪЪгУгкХфЦНЫЎШмвКжаЕФЗДгІЁЃ
бѕЛЏЛЙдЗДгІХфЦНгІеЦЮевдЯТЛљБОвЊЧѓЃКЃЈ1ЃЉбѕЛЏМСКЭЛЙдМСЕФбѕЛЏжЕБфЛЏБиаыЯрЕШЁЃЃЈ2ЃЉЗНГЬЪНСНБпЕФИїжждЊЫиЕФдзгЪ§БиаыЯрЕШЁЃЃЈ3ЃЉХфЦНЗНЗЈЕФФбЕуЪЧЮДЗЂЩњбѕЛЏжЕБфЛЏЕФдзгЪ§ЕФХфЦНЃЌД§Б№ЪЧЧтКЭбѕдзгЕФХфЦНЁЃвЛАуЧщПіЯТШчЗДгІЮябѕдзгЪ§ЖрСЫЃЌдкЫсадНщжЪжаЕФЗДгІвдМгH+РызгЩњГЩЫЎЕФЗНЪНХфЦНЃЛШєЮЊМюадНщжЪжаЕФЗДгІЃЌгІМгH2OЪЙжЎгыбѕЗДгІЩњГЩOHвЛРызгЁЃ
вЛАуЯШХфЦНHЁЂOвдЭтЕФдзгЪ§ЃЌШЛКѓХфЦНHЁЂOдзгЪ§ЃЌзюКѓХфЦНЕчзгЪ§
ЧыЭЌбЇУЧзЂвтЃКЫсадНщжЪжаХфЦНЕФАыЗДгІЗНГЬЪНРяВЛгІГіЯжOHЈCЃЌдкМюадНщжЪжаХфЦНЕФАыЗДгІВЛгІГіЯжH+ЁЃ
дкЧАУцЕФЬжТлжаЃЌЮвУЧНЋбѕЛЏЛЙдЗДгІЗжГЩСНИіАыЗДгІЁЃетжжУшЪіВЛНіНіЪЧХфЦНЗДгІЗНГЬЪНЕФвЛжжЗНЗЈЃЌИќживЊЕФЃЌПЩвдЪЙбѕЛЏАыЗДгІКЭЛЙдАыЗДгІдквЛЖЈзАжУжаИєРыПЊРДЃЌЗжБ№ЗЂЩњЗДгІЃЌБуПЩВњЩњЕчСїЃЌДгЖјНЋЛЏбЇФмзЊЛЏЮЊЕчФмЁЃРћгУбѕЛЏЛЙдЗДгІЃЌНЋЛЏбЇФмзЊБфЮЊЕчФмЕФзАжУНазідЕчГи(primary cell)ЁЃЕчГиЕФЩшМЦжЄУїСЫбѕЛЏЛЙдЗДгІШЗЪЕЗЂЩњСЫЕчзгЕФзЊвЦЁЃ
вЛИідЕчГиАќРЈСНИіАыЕчГиЃЌУПИіАыЕчГигжГЦЮЊвЛИіЕчМЋЁЃЦфжаЗХГіЕчзгЕФвЛМЋГЦЮЊИКМЋЃЈnegative electrodeЃЉЃЌЪЧЕчзгСїГіМЋЃЌЗЂЩњбѕЛЏЗДгІЃЛСэвЛМЋЪЧНгЪмЕчзгЕФвЛМЋГЦЮЊе§МЋЃЈpositive electrodeЃЉЃЌе§МЋЩЯЗЂЩњЛЙдЗДгІЁЃЕчМЋЩЯЗжБ№ЗЂЩњЕФбѕЛЏЛЙдЗДгІЃЌГЦЮЊЕчМЋЗДгІЃЈelectrode reactionЃЉЁЃвЛАуЫЕРДЃЌгЩСНжжН№ЪєЕчМЋЙЙГЩЕФдЕчГиЃЌНЯЛюЦУЕФН№ЪєзіИКМЋЃЌСэвЛН№Ъєзіе§МЋЁЃИКМЋН№ЪєЪЇШЅЕчзгГЩЮЊРызгЖјНјШыШмвКЃЌЫљвдЫќзмЪЧж№НЅШмНтЁЃ
дЕчГиЕФСНИіЕчМЋЕФШмвКЭЈЙ§бЮЧХЙЕЭЈЃЌбЮЧХгаСНЗНУцЕФзїгУЃЌвЛЗНУцЫќПЩвдЯћГ§вђШмвКжБНгНгДЅЖјаЮГЩЕФвКНгЕчЪЦЃЌСэвЛЗНУцЫќПЩЪЙСЊНгЕФСНШмвКБЃГжЕчжаадЃЌР§ШчЃЌCuЈDZnдЕчГиЃКZn+Cu2+=Cu+Zn2+
дЕчМЋе§МЋЗЂЩњЛЙдЗДгІЃЌИКМЋЗЂЩњбѕЛЏЗДгІ
ИКМЋЃКZnЃ2e=Zn2+ЃЈбѕЛЏЬЌЩ§ИпЃЉ
е§МЋЃКCu2++2e=CuЃЈбѕЛЏЬЌНЕЕЭЃЉ
дЕчГиЕФЗћКХЃКЮЊБэДяЗНБуЭЈГЃНЋдЕчГиЕФзщГЩвдЙцЖЈЕФЗНЪНЪщаДЃЌГЦЮЊЕчГиЗћКХБэЪОЪНЁЃЦфЪщаДддђЙцЖЈЃК
ЃЈ1ЃЉАбИКМЋаДдкЕчГиЗћКХБэЪОЪНЕФзѓБпЃЌвдЁАЃЈЃЃЉЁББэЪОЃЛе§МЋаДдкЕчГиЗћКХБэЪОЪНЕФгвБпЃЌВЂвдЁАЃЈ+ЃЉЁББэЪОЁЃ
ЃЈ2ЃЉвдЛЏбЇЪНБэЪОЕчГижаИїЮяжЪЕФзщГЩЃЌШмвКвЊБъЩЯХЈЖШЛђЛюЖШЃЈmol.L-1ЃЉЃЌШєЮЊЦјЬхЮяжЪгІзЂУїЦфЗжбЙЃЈPaЃЉЁЃШчВЛЬиЪтжИУїЃЌдђЮТЖШЮЊ298KЃЌЦјЬхЗжбЙЮЊ101.325kPaЃЌШмвКХЈЖШЮЊ1 mol.L-1ЁЃ
ЃЈ3ЃЉвдЗћКХЁА|ЁББэЪОВЛЭЌЮяЯржЎМфЕФНгНчЃЌгУЁАЁЌЁББэЪОбЮЧХЁЃЭЌвЛЯржаЕФВЛЭЌЮяжЪжЎМфгУЁАЃЌЁББэЪОЁЃ
ЃЈ4ЃЉЗЧН№ЪєЛђЦјЬхВЛЕМЕчЃЌвђДЫЗЧН№ЪєдЊЫидкВЛЭЌМлЬЌЪБЙЙГЩЕФбѕЛЏЛЙдЕчЖдзїАыЕчГиЪБЃЌашЭтМгЖшадН№ЪєЃЈШчВЌКЭЪЏФЋЕШЃЉзіЕчМЋЕМЬхЁЃЦфжаЃЌЖшадН№ЪєВЛВЮгыЗДгІЃЌжЛЦ№ЕМЕчЕФзїгУЁЃ
ЕчГие§ЁЂИКЕчМЋжЎМфУЛгаЕчСїЭЈЙ§ЪБЕФЕчЪЦВюГЦЮЊЕчГиЕФЕчЖЏЪЦЃЈelectronmotiveforeЗћКХEГиБэЪОЃЉЁЃЕчГиЕчЖЏЪЦЪЧКтСПбѕЛЏЛЙдЗДгІЭЦЖЏСІДѓаЁЕФХаОнЃЌетгыШШСІбЇЩЯЪЙгУЗДгІЬхЯЕЕФМЊВМЫЙздгЩФмБфЛЏІЄGзїЮЊЗДгІздЗЂЧуЯђЕФХаОнЪЧвЛжТЕФЁЃ
EГи=EЃЈ+ЃЉЃEЃЈ-ЃЉ
ЕчГиЕчЖЏЪЦЪЧЕчГиЗДгІНјааЕФЭЦЖЏСІЁЃЕБгЩбѕЛЏЛЙдЗДгІЙЙГЩЕФЕчГиЕФЕчЖЏЪЦEІеГиДѓгкСуЪБЃЌдђДЫбѕЛЏЛЙдЗДгІОЭФмздЗЂНјааЁЃвђДЫЃЌЕчГиЕчЖЏЪЦвВЪЧХаЖЯбѕЛЏЛЙдЗДгІФмЗёздЗЂНјааЕФХаОнЁЃ
ЕчГиЭЈЙ§бѕЛЏЛЙдЗДгІВњЩњЕчФмЃЌЬхЯЕЕФздгЩФмНЕЕЭЁЃдкКуЮТКубЙЯТЃЌздгЩФмЕФНЕЕЭжЕЃЈЃЁїGЃЉЕШгкЕчГиПЩФмзїГіЕФзюДѓгагУЕчЙІЃЈWЕчЃЉЃКЃЈдкЕчГиЗДгІжаЃЌШчЙћЗЧХђеЭЙІжЛгаЕчЙІвЛжжЃЉЃЌгЩЕчЙІ=ЕчКЩСП*ЕчЪЦВюЃЌЕУЃК
ЃЁїGЃНWЕчЃНQEЃНnFEГи
МДЁїGЃНЃnFEГи
дкБъзМзДЬЌЯТЃЌЩЯЪНПЩаДГЩЃК
ЁїGІеЃНЃnFEІеГи
ЕБEІеГиЮЊе§жЕЪБЃЌЁїGІеЮЊИКжЕЃЌдкБъзМзДЬЌЯТбѕЛЏЛЙдЗДгІе§ЯђздЗЂНјааЃЛЕБEІеГиЮЊИКжЕЪБЃЌЁїGІеЮЊе§жЕЃЌдкБъзМзДЬЌЯТЗДгІе§ЯђЗЧздЗЂНјааЃЌФцЯђЗДгІздЗЂНјааЁЃEЛђEІегњЪЧНЯДѓЕФе§жЕЃЌбѕЛЏЛЙдЗДгІе§ЯђздЗЂНјааЕФЧуЯђгњДѓЁЃEГиЛђEІеГигњЪЧНЯДѓЕФИКжЕЃЌФцЯђЗДгІздЗЂНјааЕФЧуЯђгњДѓЁЃ
ЯТУцЮвУЧНщЩмЃЌБъзМЧтЕчМЋКЭИЪЙЏЕчМЋЕФЕчМЋЕчЪЦЃК
БъзМЧтЕчМЋЪЧвЛжжМйЖЈЕФРэЯызДЬЌЃЌЭЈГЃЪЧНЋЖЦгавЛВуКЃУрзДВЌКкЕФВЌЦЌЃЌНўШыЕНHХЈЖШЮЊ1.0mol/LЕФЫсШмвКжаЃЌВЛЖЯЭЈШыбЙСІЮЊ100kPaЕФДПЧтЦјЃЌЪЙВЌКкЮќИНH2жСБЅКЭЃЌетЪБВЌЦЌОЭКУЯёЪЧгУЧтжЦГЩЕФЕчМЋвЛбљЁЃЦфЕчМЋЗћКХЃКPt|H2(101.3kPa)|H+(1mol.L-1)
ЕчМЋЗДгІЃК2H++2e=H2ЃЈgЃЉ
ЮвУЧЙцЖЈЃЌБъзМЧтЕчМЋЕФЛЙдЕчМЋЕчЪЦЮЊСуЃЌМДEІеH+/H2ЃН0V
гвЩЯНЧЕФЗћКХЁАІеЁБДњБэБъзМЬЌЁЃ
БъзМЬЌвЊЧѓЕчМЋДІгкБъзМбЙСІЃЈ101.325kPaЃЉЯТЃЌзщГЩЕчМЋЕФЙЬЬхЛђвКЬхЮяжЪЖМЪЧДПОЛЮяжЪЃЛЦјЬхЮяжЪЦфЗжбЙЮЊ101.325kPaЃЛзщГЩЕчЖдЕФгаЙиРызгЃЈАќРЈВЮгыЗДгІЕФНщжЪЃЉЕФХЈЖШЮЊ1mol.L-1ЃЈбЯИёЕФИХФюЪЧЛюЖШЃЉЁЃЭЈГЃВтЖЈЕФЮТЖШЮЊ298KЁЃ
гЩгкБъзМЧтЕчМЋЕФжЦзїКЭЪЙгУЖМКмРЇФб,ЦНЪБШЫУЧВЩгУЯрЖдЮШЖЈЕФИЪЙЏЕчМЋзїВЮБШЕчМЋ. БЅКЭИЪЙЏЕчМЋЃЈSCEЃЉЕчМЋНсЙЙШчЭМЫљЪОЁЃЕчМЋгЩСНИіВЃСЇЬзЙмзщГЩЃЌФкЙмЩЯВПЮЊЙЏЃЌСЊгаPtЫПзїЕчМЋв§ЯпЃЌжаВПЮЊHg2Cl2ЃHgК§зДЮяЃЌЕзВПгУЫиЩеДЩШћНєЃЌЭтЙмЯТВПжЇЙмПкШћгаЖрПзЫиЩеДЩЃЌЩЯВПВрПкзЂШыБЅКЭKClШмвКЃЌВтЖЈжаЪЂгаKClБЅКЭШмвКЕФЭтЙмЛЙПЩЦ№ЕНбЮЧХЕФзїгУЁЃИЪЙЏЕчМЋЕчМЋЗћКХЃКHgЃЈlЃЉ|Hg2Cl2ЃЈsЃЉ|KClЃЈcЃЉ
ЕчМЋЗДгІЪНЃКHg2Cl2ЃЋ2e=2HgЃЋ2Cl-
вдБъзМЧтЕчМЋЕФЛЙдЕчМЋЕчЪЦЮЊЛљзМЃЌПЩвдВтЕУБъзМИЪЙЏЕчМЋЕФЕчЪЦЮЊ0.2415VЁЃ
ЮЊСЫЛёЕУИїжжЕчМЋЕФЕчМЋЕчЪЦЪ§жЕЃЌЭЈГЃвдБъзМЕчМЋКЭД§ВтЕчМЋдкБъзМзДЬЌЯТзщГЩЕчГиЃЌВтЕУИУЕчГиЕФЕчЖЏЪЦжЕЃЌВЂЭЈЙ§жБСїЕчбЙБэШЗЖЈЕчГиЕФе§ИКМЋЃЌМДПЩИљОнEГи=EЃЈ+ЃЉЃEЃЈ-ЃЉМЦЫуИїжжЕчМЋЕФБъзМЕчМЋЕчЪЦЕФЯрЖдЪ§жЕЁЃНЋВЛЭЌбѕЛЏЛЙдЕчЖдЕФБъзМЕчМЋЕчЪЦЪ§жЕАДеегЩаЁЕНДѓЕФЫГађХХСаЃЌЕУЕНЕчМЋЗДгІЕФБъзМЕчМЋЕчЪЦБэЁЃЦфЬиЕугаЃК
ЃЈlЃЉвЛАуВЩгУЕчМЋЗДгІЕФЛЙдЕчЪЦЃЌУПвЛЕчМЋЕФЕчМЋЗДгІОљаДГЩЛЙдЗДгІаЮЪНЃЌМДЃК
бѕЛЏаЭЃЋne=ЛЙдаЭЃЌ
ЃЈ2ЃЉБъзМЕчМЋЕчЪЦЪЧЦНКтЕчЪЦЃЌУПИіЕчЖдEІежЕЕФе§ИККХЃЌВЛЫцЕчМЋЗДгІНјааЕФЗНЯђЖјИФБфЁЃ
ЃЈ3ЃЉEІежЕЕФДѓаЁПЩгУвдХаЖЯдкБъзМзДЬЌЯТЕчЖджабѕЛЏаЭЮяжЪЕФбѕЛЏФмСІКЭЛЙдаЭЮяжЪЕФЛЙдФмСІЕФЯрЖдЧПШѕЃЌЖјгыВЮгыЕчМЋЗДгІЮяжЪЕФЪ§СПЮоЙи,ЮоМгКЭадЁЃР§ШчЃК
I2ЃЋ2e=2I-ЃЌEІе=+0.5355V
1/2I2ЃЋe=I-ЃЌEІе=+0.5355V
ЃЈ4ЃЉEІежЕНіЪЪКЯгкБъзМЬЌЪБЕФЫЎШмвКЪБЕФЕчМЋЗДгІЁЃЖдгкЗЧЫЎЁЂИпЮТЁЂЙЬЯрЗДгІЃЌдђВЛЪЪКЯЁЃ
ЕчМЋЕчЪЦЕФДѓаЁГ§СЫШЁОігкЕчЖдЕФБОадЭтЃЌЛЙЪмЕНЫќУЧЕФХЈЖШЃЈЛђЦјЬхЕФЗжбЙЃЉКЭЮТЖШЕФгАЯьЁЃБъзМЕчМЋЕчЪЦЪЧдкБъзМзДЬЌЯТВтЖЈЕФЃЌЕЋЪЧЖдгкОјДѓЖрЪ§ЕФбѕЛЏЛЙдЗДгІРДЫЕЃЌВЂЗЧдкБъзМзДЬЌЯТНјааЕФЃЌДгЖјЪЙЕчЖдЕФЕчМЋЕчЪЦвВЫцжЎЗЂЩњИФБфЁЃЕчМЋЕчЪЦгыХЈЖШЃЈЛђбЙСІЃЉЁЂЮТЖШМфЖЈСПЙиЯЕПЩгЩФмЫЙЬиЃЈNernstЃЉЗНГЬЪНБэЪОЃК
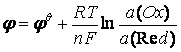
етИіЙиЯЕЪНГЦЮЊNernstЗНГЬЪНЃЌЫќЗДгГСЫВЮМгЕчМЋЗДгІЕФИїЮяжЪЕФХЈЖШвдМАЮТЖШЖдЕчМЋЕчЪЦЕФгАЯьЁЃЪНжа![]() ЪЧбѕЛЏаЭЮяжЪКЭЛЙдаЭЮяжЪЮЊШЮвтХЈЖШЪБЕчЖдЕФЕчМЋЕчЪЦЃЛ
ЪЧбѕЛЏаЭЮяжЪКЭЛЙдаЭЮяжЪЮЊШЮвтХЈЖШЪБЕчЖдЕФЕчМЋЕчЪЦЃЛ![]() ЪЧЕчЖдЕФБъзМЕчМЋЕчЪЦЃЌRЪЧФІЖћЦјЬхГЃЪ§ЃЌЕШгк8.314KJЁЄmol-1ЁЄk-1ЃЛFЮЊFaradayГЃЪ§ЃЈ96485CЁЄmol-1ЃЉЃЛTЮЊШШСІбЇЮТЖШЃЈKЃЉЃЛnЮЊЕчМЋЗДгІЕФЕчзгЪ§ЃЛaЃЈOxЃЉКЭaЃЈRedЃЉЗжБ№БэЪОЕчМЋЗДгІЪНжабѕЛЏаЭЮяжЪКЭЛЙдаЭЮяжЪЕФЛюЖШЁЃШчЙћЪЧЯЁШмвКЃКa=cЃЛШчЙћЪЧбЙСІНЯЕЭЕФЦјЬхЃК
ЪЧЕчЖдЕФБъзМЕчМЋЕчЪЦЃЌRЪЧФІЖћЦјЬхГЃЪ§ЃЌЕШгк8.314KJЁЄmol-1ЁЄk-1ЃЛFЮЊFaradayГЃЪ§ЃЈ96485CЁЄmol-1ЃЉЃЛTЮЊШШСІбЇЮТЖШЃЈKЃЉЃЛnЮЊЕчМЋЗДгІЕФЕчзгЪ§ЃЛaЃЈOxЃЉКЭaЃЈRedЃЉЗжБ№БэЪОЕчМЋЗДгІЪНжабѕЛЏаЭЮяжЪКЭЛЙдаЭЮяжЪЕФЛюЖШЁЃШчЙћЪЧЯЁШмвКЃКa=cЃЛШчЙћЪЧбЙСІНЯЕЭЕФЦјЬхЃК![]() ЃЛШчЙћЪЧЙЬЬхЛђДПвКЬхЃКa=1ЃЌдйNernstЗНГЬЪНжаПЩвдВЛСаШыЁЃСэЭтЃЌЛюЖШЕФЗНДЮЕШгкИУЮяжЪдкЕчМЋЗДгІжаЕФЛЏбЇМЦСПЪ§ЁЃ
ЃЛШчЙћЪЧЙЬЬхЛђДПвКЬхЃКa=1ЃЌдйNernstЗНГЬЪНжаПЩвдВЛСаШыЁЃСэЭтЃЌЛюЖШЕФЗНДЮЕШгкИУЮяжЪдкЕчМЋЗДгІжаЕФЛЏбЇМЦСПЪ§ЁЃ
ЕБЮТЖШЮЊ298.15KЪБЃЌНЋИїГЃЪ§ДјШыЩЯЪНжаЃЌВЂНЋздШЛЖдЪ§ЛЛГЩГЃгУЖдЪ§ЃЌМДЕУЃК
![]()
дкгаH+ЛђOHЃРызгВЮгыЕФЕчМЋЗДгІжаЃЌЫсЖШЕФИФБфЛсгАЯьЕчМЋЕчЪЦЁЃР§ШчжиИѕЫсМиЕФбѕЛЏадЃЌЛсЫцзХЫсЖШЕФБфЛЏЖјИФБфЁЃЦфЕчМЋЗДгІЮЊЃК
K2Cr2O7ЃЋ14H+ЃЋ6e![]() 2Cr3+ЃЋ7H2OЃЌEІеCr2O72-/Cr3+ЃН1.33ЃЈVЃЉ
2Cr3+ЃЋ7H2OЃЌEІеCr2O72-/Cr3+ЃН1.33ЃЈVЃЉ
ЗДгІжаКЌгаH+ЃЌШєНЋ[Cr2O72-]КЭ[Cr3+]ЖМЖЈЮЊ1 mol.LЃ1ЪБЃЌгЩФмЫЙЭбЗНГЬЪНПЩвдПДГіНщжЪЕФЫсЖШЖдЕчМЋЕчЪЦЕФгАЯьЁЃ
E=EІеCr2O72-/Cr3+ЃЋ0.0592/6lg[H+]14
K2Cr2O7ЕФбѕЛЏадЫцзХЫсЖШдіЧПЖјЯджјдіМгЃЛЖјЫцзХЫсЖШЕФМѕШѕЖјЯджјМѕШѕЁЃвђЮЊИУЕчМЋЗДгІЧтРызгХЈЖШжИЪ§КмИпЃЌШмвКЫсЖШБфЛЏЖдЕчМЋЕчЪЦЕФгАЯьЃЌдЖдЖНЯбѕЛЏаЭКЭЛЙдаЭЮяжЪБОЩэХЈЖШБфЛЏв§Ц№ЕФгАЯьДѓЕУЖрЁЃЙЪдкЪЙгУK2Cr2O7ЁЂKMnO4ЕШКЌбѕЫсбЮзїЮЊбѕЛЏМСЪБЃЌзмЪЧвЊНЋШмвКЫсЛЏЃЌвдБЃГжЫсадНщжЪжаГфЗжЗЂЛгИУРрбѕЛЏМСЕФбѕЛЏадЁЃ
ФбШмЛЏКЯЮяЁЂХфКЯЮяЕФаЮГЩЖдЕчМЋЕчЪЦЕФгАЯьЃКдкЗДгІжаМгШыГСЕэМСЪЙбѕЛЏЬЌЮяжЪЛђЛЙдЬЌЮяжЪзЊБфГЩГСЕэЃЌПЩДѓДѓНЕЕЭбѕЛЏЬЌЮяжЪЛђЛЙдЬЌЮяжЪЕФХЈЖШЃЌДгЖјЕМжТЕчМЋЕчЪЦЗЂЩњКмДѓЕФБфЛЏЁЃШчдкЕчМЋЗДгІAg+ЃЋe=AgЬхЯЕжаМгШыТШЛЏФЦКѓЃЌгЩгкЗЂЩњAg++Cl-AgClГСЕэЗДгІЃЌжТЪЙШмвКжаЃлAg+ЃнРызгХЈЖШНЕЕЭЃЌИљОнГСЕэШмЖШЛ§дРэЃЌЗДгІДяЕНЦНКтКѓЃЌШє[Cl-]=1mol.L-1ЃлAg+ЃнРызгХЈЖШЮЊЃК
ЃлAg+ЃнЃНЃЫsp/[Cl-ЃнЃН1.8ЁС10-10(mol.LЃ1)
етЪБAg+/AgЕчЖдЕФЕчМЋЕчЪЦЮЊЃК
EAg+/AgЃНEІеAg+/AgЃЋ0.0592lg[Ag+]
ЃН0.7996ЃЋ0.0592lg[1.8ЁС10-10]ЃН0.2227ЃЈVЃЉ
ЫљвдЃЌгЩгкAgClГСЕэЕФаЮГЩЃЌвјЕчМЋЕчЪЦДг0.7996VЯджјЕиЯТНЕЮЊ0.2227VЃЌМДAg+ЕФбѕЛЏадДѓДѓМѕШѕСЫЁЃдкН№ЪєЕчМЋЬхЯЕжааЮГЩХфКЯЮяЪБЃЌвВНЋЪЙЕчМЋЕчЪЦНЕЕЭЃЌбѕЛЏЬЌЕФбѕЛЏФмСІМѕШѕЃЌЖјН№ЪєЕФЛЙдФмСІдіЧПЁЃ
ЕчМЋЕчЪЦЕФгІгУЃКдкБъзМзДЬЌЯТбѕЛЏМСКЭЛЙдМСЕФЯрЖдЧПШѕЃЌПЩжБНгБШНЯEІежЕЕФДѓаЁЁЃEІежЕНЯаЁЕФЕчМЋЦфЛЙдаЭЮяжЪгњвзЪЇШЅЕчзгЃЌЪЧгњЧПЕФЛЙдМСЃЌЖдгІЕФбѕЛЏаЭЮяжЪдђгњФбЕУЕНЕчзгЃЌЪЧгњШѕЕФбѕЛЏМСЁЃEІежЕгњДѓЕФЕчМЋЦфбѕЛЏаЭЮяжЪгњвзЕУЕНЕчзгЃЌЪЧНЯЧПЕФбѕЛЏМСЃЌЖдгІЕФЛЙдаЭЮяжЪдђгњФбЪЇШЅЕчзгЃЌЪЧгњШѕЕФЛЙдМСЁЃдкБъзМЕчМЋЕчЪЦБэжаЃЌЛЙдаЭЕФЛЙдФмСІздЩЯЖјЯТвРДЮМѕШѕЃЌбѕЛЏаЭЕФбѕЛЏФмСІздЩЯЖјЯТвРДЮдіЧПЁЃ
ИљОнEІежЕЃЌХаЖЯБъзМзДПіЯТбѕЛЏЛЙдЗДгІНјааЕФЗНЯђЃКЭЈГЃЬѕМўЯТЃЌбѕЛЏЛЙдЗДгІзмЪЧгЩНЯЧПЕФбѕЛЏМСгыЛЙдМСЯђзХЩњГЩНЯШѕЕФбѕЛЏМСКЭЛЙдМСЗНЯђНјааЁЃДгЕчМЋЕчЪЦЕФЪ§жЕРДПДЃЌЕБбѕЛЏМСЕчЖдЕФЕчЪЦДѓгкЛЙдМСЕчЖдЕФЕчЪЦЪБЃЌЗДгІВХПЩвдНјааЁЃЗДгІвдЁАИпЕчЪЦЕФбѕЛЏаЭбѕЛЏЕЭЕчЪЦЕФЛЙдаЭЁБЕФЗНЯђНјааЁЃдкХаЖЯбѕЛЏЛЙдЗДгІФмЗёздЗЂНјааЪБЃЌЭЈГЃжИЕФЪЧе§ЯђЗДгІЁЃ
ИљОнEІеГижЕЃЌХаЖЯбѕЛЏЛЙдЗДгІНјааЗНЯђЃКЕчГиЕчЖЏЪЦЪЧЕчГиЗДгІНјааЕФЭЦЖЏСІЁЃЕБгЩбѕЛЏЛЙдЗДгІЙЙГЩЕФЕчГиЕФЕчЖЏЪЦEІеГиДѓгкСуЪБЃЌдђДЫбѕЛЏЛЙдЗДгІОЭФмздЗЂНјааЁЃвђДЫЃЌЕчГиЕчЖЏЪЦвВЪЧХаЖЯбѕЛЏЛЙдЗДгІФмЗёздЗЂНјааЕФХаОнЁЃЕчГиЭЈЙ§бѕЛЏЛЙдЗДгІВњЩњЕчФмЃЌЬхЯЕЕФздгЩФмНЕЕЭЁЃдкКуЮТКубЙЯТЃЌздгЩФмЕФНЕЕЭжЕЃЈЃЁїGЃЉЕШгкЕчГиПЩФмзїГіЕФзюДѓгагУЕчЙІЃЈWЕчЃЉЃК
ЃЁїGЃНWЕчЃНQEЃНnFEГи
МДЁїGЃНЃnFEГи
дкБъзМзДЬЌЯТЃЌЩЯЪНПЩаДГЩЃК
ЁїGІеЃНЃnFEІеГи
ЕБEІеГиЮЊе§жЕЪБЃЌЁїGІеЮЊИКжЕЃЌдкБъзМзДЬЌЯТбѕЛЏЛЙдЗДгІе§ЯђздЗЂНјааЃЛЕБEІеГиЮЊИКжЕЪБЃЌЁїGІеЮЊе§жЕЃЌдкБъзМзДЬЌЯТЗДгІе§ЯђЗЧздЗЂНјааЃЌФцЯђЗДгІздЗЂНјааЁЃEЛђEІегњЪЧНЯДѓЕФе§жЕЃЌбѕЛЏЛЙдЗДгІе§ЯђздЗЂНјааЕФЧуЯђгњДѓЁЃEГиЛђEІеГигњЪЧНЯДѓЕФИКжЕЃЌФцЯђЗДгІздЗЂНјааЕФЧуЯђгњДѓЁЃ
ИљОнEІеГижЕЃЌШЗЖЈЗДгІНјааЕФЯоЖШЁЊЁЊМЦЫуЦНКтГЃЪ§ЃКвЛИіЛЏбЇЗДгІЕФЭъГЩГЬЖШПЩДгИУЗДгІЕФЦНКтГЃЪ§ДѓаЁЖЈСПЕиХаЖЯЁЃвђДЫЃЌАбБъзМЦНКтГЃЪ§KІеКЭШШСІбЇМЊВМЫЙздгЩФмСЊЯЕЦ№РДЁЃ
RЮЊЦјЬхГЃЪ§ЃЌTЮЊОјЖдЮТЖШЃЌnЮЊбѕЛЏЛЙдЗДгІЗНГЬжаЕчзгзЊвЦЪ§ФПЃЌFЮЊЗЈРЕкГЃЪ§ЁЃИУЪНБэУїЃЌдквЛЖЈЮТЖШЯТЃЌбѕЛЏЛЙдЗДгІЕФЦНКтГЃЪ§гыБъзМЕчГиЕчЖЏЪЦгаЙиЃЌгыЗДгІЮяЕФХЈЖШЮоЙиЁЃEІедНДѓЃЌЦНКтГЃЪ§ОЭдНДѓЃЌЗДгІНјаадНЭъШЋЁЃвђДЫЃЌПЩвдгУEІежЕЕФДѓаЁРДЙРМЦЗДгІНјааЕФГЬЖШЁЃвЛАуЫЕЃЌEІеЁн0.2ЁЋ0.4ЃжЕФбѕЛЏЛЙдЗДгІЃЌЦфЦНКтГЃЪ§ОљДѓгк106(K>106),БэУїЗДгІНјааЕФГЬЖШвбЯрЕБЭъШЋСЫЁЃKІежЕДѓаЁПЩвдЫЕУїЗДгІНјааЕФГЬЖШЃЌЕЋВЛФмОіЖЈЗДгІЫйТЪЁЃ
ЕБФГдЊЫиПЩвдаЮГЩШ§жжЛђШ§жжвдЩЯбѕЛЏжЕЕФЮяжжЪБЃЌетаЉЮяжжПЩвдзщГЩЖржжВЛЭЌЕФЕчЖдЃЌИїЕчЖдЕФБъзМЕчМЋЕчЪЦПЩгУЭМЕФаЮЪНБэЪОГіРДЃЌетжжЭМНазідЊЫиЕчЪЦЭМЁЃЛдЊЫиЕчЪЦЭМЪБЃЌПЩвдАДдЊЫиЕФбѕЛЏжЕгЩИпЕНЕЭЕФЫГађЃЌАбИїЮяжжЕФЛЏбЇЪНДгзѓЕНгваДГіРДЃЌИїВЛЭЌбѕЛЏжЕЮяжжжЎМфгУжБЯпСЌНгЦ№РДЃЌдкжБЯпЩЯБъУїСНжжВЛЭЌбѕЛЏжЕЮяжжЫљзщГЩЕФЕчЖдЕФБъзМЕчМЋЕчЪЦЁЃдЊЫиЕчЪЦЭМЪЧСЫНтвЛжждЊЫиЕФЖржжбѕЛЏЬЌЕФЮяжЪжЎМфБфЛЏЙиЯЕЕФвЛжжБэЪОЗНЗЈЁЃдЊЫиЕчЪЦЭМгжПЩЗжЮЊЫсадШмвКЕчЪЦЭМКЭМюадШмвКЕчЪЦЭМЁЃ
дЊЫиЕчЪЦЭМжївЊгІгУЃК
1ЃЎХаЖЯЦчЛЏЗДгІЪЧЗёФмНјаа
ЫљЮНЦчЛЏЗДгІЃЌОЭЪЧдкЭЌвЛИідЊЫижаЃЌвЛВПЗждзгЃЈЛђРызгЃЉБЛбѕЛЏЃЌСэвЛВПЗждзгЃЈЛђРызгЃЉБЛЛЙдЕФЗДгІЁЃШєдкЯТСадЊЫиЕчЪЦЭМжа
EІезѓEІегв
AЁЊЁЊBЁЊЁЊC
ШєEІегвЃОEІезѓ,ЦфжаМфМлЬЌBПЩздЗЂЕиЗЂЩњсЊЛЏЗДгІ,ЩњГЩAКЭCЁЃЧвEІеГидНДѓЃЌЦчЛЏЗДгІГЬЖШдНДѓЁЃЯрЗДЕиЃЌШєEІегвЃМEІезѓЃЌдђВЛФмЗЂЩњЦчЛЏЗДгІЃЌЗЂЩњЦчЛЏЗДгІЕФФцЗДгІЁЃ
2ЃЎМЦЫуБъзМЕчМЋЕчЪЦ
ИљОндЊЫиЕчЪЦЭМПЩДгМИИіЯрСкбѕЛЏЬЌЕчЖдЕФвбжЊБъзМЕчМЋЕчЪЦЃЌПЩвдЧѓЫуВЛЯрСкбѕЛЏЬЌЕчЖдЕФЮДжЊБъзМЕчМЋЕчЪЦЁЃ
аЁНсЃЌдкБОеТжаЃЌЮвУЧбЇЯАСЫЕчЛЏбЇЛљДЁжЊЪЖЁЃдЕчГиЕчЖЏЪЦЃЌдЕчГиЕФзюДѓЙІгыGibssКЏЪ§ЃЌNernstЗНГЬЪНЃЌХаЖЯбѕЛЏЛЙдЗДгІЗНЯђЃЌШЗЖЈбѕЛЏЛЙдЗДгІЕФЯоЖШЃЌЪЧБОеТЕФжиЕуМАФбЕуЃЌЧыЭЌбЇУЧНсКЯПЮКѓЯАЬтЃЌЖдЫљбЇжЊЪЖМгвдЙЎЙЬЁЃ
ЕкЮхеТ дзгНсЙЙгыдЊЫижмЦкад
ЭЌбЇУЧКУЃЌДгНёЬьПЊЪМЃЌЮвУЧНјШыБОПЮГЬЕФЕкЖўВПЗжЃЌЮяжЪНсЙЙЛљДЁЁЃдкЕквЛВПЗжжаЃЌЮвУЧвбОЬжТлСЫЛЏбЇЗДгІЕФЗНЯђЃЌЯоЖШЕШЮЪЬтЃЌетаЉЬжТлЖМЪЧдкКъЙлВуУцЩЯЃЌЖдДѓСПЕФЗжзгааЮЊНјааСЫЬжТлЁЃдкетВПЗжжаЃЌЮвУЧвЊНјШыЮЂЙлВуУцЁЃЮвУЧжЊЕРЃЌдзгЭЈЙ§ИФБфЫќУЧЕФНсКЯЗНЪНЃЌДгвЛжжЗжзгБфГЩСэвЛжжЗжзгЃЌЕЋетжжНсКЯЗНЪНЕФИФБфДгБОжЪЩЯЪЧЕчзгНсЙЙЕФБфЛЏЃЌЫљвдЃЌЮвУЧдкетвЛеТжаЃЌвЊДгдзгЕФЕчзгНсЙЙПЊЪМЬжТлЁЃ
дкЬжТлЕФдзгЕФЕчзгНсЙЙжЎЧАЃЌЮвУЧгаБивЊЛиЙЫвЛЯТМИИіживЊЕФдзгНсЙЙФЃаЭЁЃ19ЪРМЭГѕЃЌгЂЙњПЦбЇМвЕРЖћЖйЬсГіНќДњдзгбЇЫЕЃЌЫћШЯЮЊдзгЪЧЮЂаЁЕФВЛПЩЗжИюЕФЪЕаФЧђЬхЃЌ100ФъКѓЃЌ1897ФъЃЌгЂЙњПЦбЇМвЬРФЗЩњЗЂЯжСЫЕчзгЃЌ1904ФъЬсГіЦЯЬбИЩУцАќЪНЕФдзгНсЙЙФЃаЭЃЌЫћШЯЮЊе§ЕчКЩЯёСїЬхвЛбљОљдШЗжВМдкдзгжаЃЌЕчзгОЭЯёЦЯЬбИЩвЛбљЩЂВМдке§ЕчКЩжаЃЌЫќУЧЕФИКЕчКЩгыФЧаЉе§ЕчКЩЯрЛЅЕжЯћЁЃ1911ФъгЂЙњЮяРэбЇМвТЌЩЊИЃЗЂЯжСЫдзгКЫЃЌНЈСЂСЫдзгКЫЪННсЙЙФЃаЭЃЌШЯЮЊдзгЕФДѓВПЗжжЪСПМЏжадквЛИіКмаЁЕФНсЙЙЩЯЃЌНадзгКЫЃЌЕчзгдкдзгКЫЭтШЦКЫзїЙьЕРдЫЖЏЃЌдзгКЫДје§ЕчЃЌЕчзгДјИКЕчЁЃ1913ФъЃЌВЈЖћЬсГіСЫЕчзгЗжВуХХВМЕФФЃаЭЃЌетЪЧвЛЛсЮвУЧвЊжиЕуЬжТлЕФФЃаЭЁЃЯждкЃЌЦеБщБЛНгЪмЕФЪЧ1926ФъЛљгкСПзгСІбЇЫљНЈСЂЕФдзгНсЙЙФЃаЭЁЃ
вЊЬжТлдзгжаЕФЕчзгНсЙЙЃЌЮвУЧвЊДгЧтдзгЙтЦзЫЕЦ№ЃЌЯждкЃЌЮвУЧвбОжЊЕРЃЌЙтЪЧвЛжжЕчДХЗјЩфЃЌЖдгкЧтдзгЕФЙтЦзбаОПБэУїЃЌдкПЩМћЙтЧјЃЌЧтдзггаЫФЬѕВЛСЌајЕФЙтЦзЯпЁЃВЛСЌајЕФЙтЦзЯпЕФЦЕТЪОпгавЛЖЈЕФЙцТЩЃЌЗћКЯетбљвЛИіОбщЙЋЪНЃЌЕЋЪЧЪНжа 2ЃЌnЃЌ3.289ЁС1015ИїДњБэЪВУДвтвхВЂВЛУїШЗЁЃ1913ФъЃЌВЈЖћдкPlanckСПзгТлКЭEinsteinЙтзгТлЕФЛљДЁЩЯЃЌЬсГіСЫаТЕФдзгНсЙЙРэТлЃЌPlanckСПзгТлЪЧжИЮЂЙлСьгђФмСПЪЧВЛСЌајЕФЁЃАЎвђЫЙЬЙЙтзгТлШЯЮЊЃЌвЛЪјЙтЪЧгЩОпгаСЃзгЬиеїЕФЙтзгЫљзщГЩЃЌУПИіЙтзгЕФФмСПгыЙтЕФЦЕТЪГЩе§БШЃЌBohrЕФРэТл(Ш§ЕуМйЩш)ЃК
ЂйКЫЭтЕчзгжЛФмдкгаШЗЖЈАыОЖКЭФмСПЕФЙьЕРЩЯдЫЖЏЃЌЧвВЛЗјЩфФмСПЃЛ
ЂкЭЈГЃЃЌЕчзгДІдкРыКЫзюНќЕФЙьЕРЩЯЃЌФмСПзюЕЭЁЊЁЊЛљЬЌЃЛдзгЛёЕУФмСПКѓЃЌЕчзгБЛМЄЗЂЕНИпФмСПЙьЕРЩЯЃЌдзгДІгкМЄЗЂЬЌЃЛ
ЂлДгМЄЗЂЬЌЛиЕНЛљЬЌЪЭЗХЙтФмЃЌЙтЕФЦЕТЪШЁОігкЙьЕРМфЕФФмСПВюЁЃ
ШчЙћЮвУЧжБЙлЕФЛГіФмМЖЭМЃЌЮвУЧОЭПЩвдПДЕУИќМгЧхГўЁЃЕЋЪЧВЈЖћЕФдзгНсЙЙРэТлЪЧЛљгкОЕфЮяРэбЇЫљЬсГіЕФЃЌгавЛЖЈВЛзужЎДІЃЌЃЈaЃЉЮоЗЈНтЪЭдзгЙтЦзЕФОЋЯИНсЙЙЃЈbЃЉВЛФмЫЕУїЖрЕчзгдзгЕФЙтЦзЃЈcЃЉВЛФмНтЪЭдзгЕФЮШЖЈадЁЃЯждкЃЌЮвУЧАбВЈЖћЕФдзгНсЙЙРэТлЖЈвхЮЊОЩСПзгТлЁЃ
ЙтЕчаЇгІЪЕбщгыАЎвђЫЙЬЙЙтзгбЇЫЕИљОнШЫУЧЕФГЃЪЖЃЌЙтЪЧвЛжжЕчДХВЈЃЌЕЋЪЧВЛФмНтЪЭЙтЕчаЇгІЕФЪЕбщЙцТЩЁЃАЎвђЫЙЬЙЪмЦеРЪПЫСПзгМйЩшЦєЗЂЃЌгк1905ФъЬсГіСЫЙтзгбЇЫЕЃКЕчДХВЈ(МДЙт)ВЛНідкКкЬхБэУцНЛЛЛФмСПЪБЃЌЪЧвЛЗнвЛЗнНјааЕФЃЌЪЧВЛСЌајЕФЃЌЖјЧвдкПеМфДЋВЅЪБЃЌвВЪЧВЛСЌајЕФЃЌЪЧвЛЗнвЛЗнЕиНјааЕФЁЃЙтВЈЪЧвЛИіИіВЛПЩЗжИюЕФСЃзгОлМЏЖјГЩЕФећЬхЁЃЙтОпгаЁАСЃзгадЁБЃЌЬхЯждкЃК
ОпгаФмСПЃК![]() ОпгаЖЏСПЃК
ОпгаЖЏСПЃК![]() ЊЋОпгажЪСПЃК
ЊЋОпгажЪСПЃК![]()
ЙтзгЕчзгЕФХізВЗўДгФмСПЪиКуКЭЖЏСПЪиКуЖЈТЩЁЃетаЉаджЪгыОЕфСІбЇжаЫљБэЪОЕФЮЂСЃаджЪЯрвЛжТЁЃетОЭЪЧАЎвђЫЙЬЙЕФЙтзгЫЕЁЃИљОнЙтЕФВЈСЃЖўЯѓадЃЌВЛНіГЩЙІЕиНтЪЭСЫЙтЕчаЇгІЕФЪЕбщЙцТЩЃЌЖјЧвЛЙНсЪјСЫМИАйФъРДЙигкЙтЪЧВЈЛЙЪЧСЃзгЕФељТлЃЌВЂЖдСПзгСІбЇЕФНЈСЂЦ№ЕНСЫОоДѓЕФДйНјзїгУЁЃ
ЕТВМТовтгк1924ФъЬсГіЮяжЪВЈМйЫЕЃКВЈСЃЖўЯѓадВЛжЛЪЧЙтВХгаЕФЬиадЃЌЖјЪЧвЛЧаЮЂЙлСЃзгЙВгаЕФБОадЁЃетвЛМйЩшдк3ФъКѓОЭЕУЕНСЫжЄЪЕЁЃ
ЕТВМТовтЙиЯЕЪНЃК![]()
МЦЫуЗЂЯжЃКжЛгаЕБЪЕЮяСЃзгЕФЕТВМТовтВЈГЄДѓгкЛђЕШгкЦфжБОЖЪБЃЌВХМШФмЯдЪОВЈЖЏадЃЌгжФмЯдЪОСЃзгадЃЌМДОпгаВЈСЃЖўЯѓадЃЛЖдгкВЈГЄаЁгкжБОЖЕФФЧаЉСЃзгЃЌСЃзгадбкИЧСЫВЈЖЏадЃЌМДжЛЯдЪОСЃзгадЁЃЬжТлБШЧтдзгЛЙДѓЕФдзг(СЃзг)ЕФВЈЖЏадвбЮоЪЕМЪвтвхЁЃДгЖјЫЕУїЃЌВЈСЃЖўЯѓадЪЧЮЂЙлСЃзгЕФЬиадЁЃ
дкЕчзгВЈЖЏадЕУЕНЪЕбщбщжЄЕФЭЌвЛФъЃЌHeisenbergЬсГіСЫВЛШЗЖЈдРэЁЃвВГЦЮЊВтВЛзМдРэЃЌетИідРэШЯЮЊЮЂЙлСЃзгЮЛжУЕФВтСПЦЋВюКЭЮЂЙлСЃзгЕФЖЏСПЦЋВюЕФГЫЛ§ВЛФмаЁгкhГ§вд4ІаЁЃЫЕУїЮЂЙлСЃзгЕФдЫЖЏВЛзёбОЕфСІбЇЕФЙцТЩЁЃЮЂЙлСЃзгЕФВЈЖЏадгыСЃзгааЮЊЕФЭГМЦадЙцТЩСЊЯЕдквЛЦ№ЃЌБэЯжЮЊЃКЮЂЙлСЃзгЕФВЈЖЏадЪЧДѓСПЮЂСЃдЫЖЏБэЯжГіРДЕФаджЪЃЌМДЪЧОпгаЭГМЦвтвхЕФИХТЪВЈЁЃ
гЩгкЮЂЙлСЃзгОпгаВЈСЃЖўЯѓадЃЌЪмВЛШЗЖЈЙиЯЕЕФЯожЦЃЌЮЂЙлСЃзгЕФдЫЖЏВЛФмгУОЕфРэТлЃЌЖјгІИУгУСПзгСІбЇЕФЗНЗЈНјааДІРэЁЃУшЪіЮЂЙлСЃзгдЫЖЏЙцТЩЕФЪ§РэЗНГЬГЦЮЊбІЖЈкЬЗНГЬЃЌЦфаЮЪНШчЯТЃК
![]()
![]() +
+![]() =-
=-![]() ЃЈЃјЃЌЃљЃЌЃњЃЉ
ЃЈЃјЃЌЃљЃЌЃњЃЉ
ЪНжа![]() ЃЈЃјЃЌЃљЃЌЃњЃЉЮЊВЈКЏЪ§ЃЌБэЪОКЫЭтЕчзгдЫЖЏзДЬЌЕФКЏЪ§ЪНЃЌЪЧЕчзгОпгаВЈЖЏадЕФБэЯжЃЛmЮЊЕчзгЕФжЪСПЃЌEЮЊЕчзгЕФзмФмСПЃЌЕШгкЖЏФмгыЪЦФмжЎКЭЃЌVЮЊЕчзгЕФЪЦФмЃЌZЮЊКЫЕчКЩЃЌeЮЊЕчзгЕчСПЃЌrЮЊЕчзгРыКЫЕФОрРыЃЌетаЉЖМЪЧЕчзгОпгаСЃзгадЕФБэЯжЃЛЃшЪЧЦеРЪПЫГЃЪ§ЃЌЭЈЙ§ЦеРЪПЫГЃЪ§ЕФСЊЯЕЃЌНЋЕчзгетжжЮЂЙлСЃзгЕФВЈЖЏадгыСЃзгадЭГвЛдкбІЖЈкЬЗНГЬжаЁЃ
ЃЈЃјЃЌЃљЃЌЃњЃЉЮЊВЈКЏЪ§ЃЌБэЪОКЫЭтЕчзгдЫЖЏзДЬЌЕФКЏЪ§ЪНЃЌЪЧЕчзгОпгаВЈЖЏадЕФБэЯжЃЛmЮЊЕчзгЕФжЪСПЃЌEЮЊЕчзгЕФзмФмСПЃЌЕШгкЖЏФмгыЪЦФмжЎКЭЃЌVЮЊЕчзгЕФЪЦФмЃЌZЮЊКЫЕчКЩЃЌeЮЊЕчзгЕчСПЃЌrЮЊЕчзгРыКЫЕФОрРыЃЌетаЉЖМЪЧЕчзгОпгаСЃзгадЕФБэЯжЃЛЃшЪЧЦеРЪПЫГЃЪ§ЃЌЭЈЙ§ЦеРЪПЫГЃЪ§ЕФСЊЯЕЃЌНЋЕчзгетжжЮЂЙлСЃзгЕФВЈЖЏадгыСЃзгадЭГвЛдкбІЖЈкЬЗНГЬжаЁЃ
бІЖЈкЬЗНГЬЪНЪЧЖўНзЦЋЮЂЗжЗНГЬЃЌЧѓНтЙ§ГЬЗЧГЃИДдгЃЌжЛаыСЫНтЦфЧѓНтЫМТЗКЭЛљБОВНжшЁЃ
ВЈКЏЪ§ЕФвтвхЃКвђЮЊ![]() 2ЃЈЃђЃЌІШЃЌІеЃЉБэЪОРыКЫОрРыЮЊЃђЃЌЗНЮЛЮЊІШЃЌІеДІЮЂаЁПеМфЕчзгдЦГіЯжЕФМИТЪУмЖШЃЌЫљвдВЈКЏЪ§
2ЃЈЃђЃЌІШЃЌІеЃЉБэЪОРыКЫОрРыЮЊЃђЃЌЗНЮЛЮЊІШЃЌІеДІЮЂаЁПеМфЕчзгдЦГіЯжЕФМИТЪУмЖШЃЌЫљвдВЈКЏЪ§![]() ЃЈЃђЃЌІШЃЌІеЃЉЪЧБэЪОКЫЭтЕчзгдЫЖЏзДЬЌЕФКЏЪ§ЃЌГЦЮЊЙьЕРКЏЪ§ЃЌМђГЦдзгЙьЕРЁЃЫфШЛЫќЪЧбІЖЈЖѕЗНГЬЕФНтЕЋШдШЛЪЧЗЧГЃИДдгЕФКЏЪ§ЪНЃЌЭЈГЃгУЭМНтЕФЗНЗЈжБЙлЕФРэНтЙьЕРКЏЪ§
ЃЈЃђЃЌІШЃЌІеЃЉЪЧБэЪОКЫЭтЕчзгдЫЖЏзДЬЌЕФКЏЪ§ЃЌГЦЮЊЙьЕРКЏЪ§ЃЌМђГЦдзгЙьЕРЁЃЫфШЛЫќЪЧбІЖЈЖѕЗНГЬЕФНтЕЋШдШЛЪЧЗЧГЃИДдгЕФКЏЪ§ЪНЃЌЭЈГЃгУЭМНтЕФЗНЗЈжБЙлЕФРэНтЙьЕРКЏЪ§![]() ЃЈЃђЃЌІШЃЌІеЃЉКЭЕчзгдЦ
ЃЈЃђЃЌІШЃЌІеЃЉКЭЕчзгдЦ![]() 2ЃЈЃђЃЌІШЃЌІеЃЉЕФЗжВМЧщПіЃЌЗжБ№ГЦЮЊЙьЕРКЏЪ§ЕФЭМаЮКЭЕчзгдЦЕФЭМаЮЁЃ
2ЃЈЃђЃЌІШЃЌІеЃЉЕФЗжВМЧщПіЃЌЗжБ№ГЦЮЊЙьЕРКЏЪ§ЕФЭМаЮКЭЕчзгдЦЕФЭМаЮЁЃ
НЋНЧЖШКЏЪ§YЃЈІШЃЌІеЃЉЖдНЧЖШЃЈІШЃЌІеЃЉзїЭМЃЌЕУдзгЙьЕРЕФНЧЖШЗжВМЭМЃЈYЭМЃЉЃЛНЋОЖЯђВЈКЏЪ§RЃЈrЃЉЖдАыОЖЃЈrЃЉзїЭМЃЌЕУдзгЙьЕРЕФОЖЯђЗжВМЭМЃЈRЭМЃЉЃЛНЋYЭМКЭRЭМЕўМгЕУдзгЙьЕРЭМаЮЁЃР§Шч2PzдзгЙьЕРЭМЁЃ
Y2ЃЈІШЃЌІеЃЉЖдНЧЖШЃЈІШЃЌІеЃЉзїЭМЃЌЕУЕчзгдЦЕФНЧЖШЗжВМЭМЃЈY2ЭМЃЉЃЛСюDЃЈrЃЉ=4![]() ЃЌдђDЃЈrЃЉЖдАыОЖЃЈrЃЉзїЭМЃЌЕУЕчзгдЦЕФОЖЯђЗжВМЭМЃЈDЭМЃЉЁЃНЋЕчзгдЦЕФНЧЖШЗжВМЭМЃЈY2ЭМЃЉгыЕчзгдЦЕФОЖЯђЗжВМЭМЃЈDЭМЃЉЕўМгЃЌБуЕУЕНЕчзгдЦЭМЁЃР§Шч2PzЕчзгдЦЭМЁЃ
ЃЌдђDЃЈrЃЉЖдАыОЖЃЈrЃЉзїЭМЃЌЕУЕчзгдЦЕФОЖЯђЗжВМЭМЃЈDЭМЃЉЁЃНЋЕчзгдЦЕФНЧЖШЗжВМЭМЃЈY2ЭМЃЉгыЕчзгдЦЕФОЖЯђЗжВМЭМЃЈDЭМЃЉЕўМгЃЌБуЕУЕНЕчзгдЦЭМЁЃР§Шч2PzЕчзгдЦЭМЁЃ
ЫФИіСПзгЪ§ЃККЫЭтЕчзгЕФСПзгЛЏЬиеїЃЌБэЯждкУшЪіЫќдЫЖЏЙцТЩЕФбІЖЈкЬЗНГЬжЛгадкЗћКЯФГаЉЬиЖЈЬѕМўЪБЃЌВХгаКЯРэЕФНт(гаШЗЖЈЕФВЈКЏЪ§)ЁЃБэЪОетаЉЬиЖЈЬѕМўЕФЮяРэСПГЦЮЊСПзгЪ§ЃЌСПзгЪ§ЪЧдкЧѓНтбІЖЈкЬЗНГЬЕФЙ§ГЬжаздШЛВњЩњЕФЁЃвЛзщСПзгЪ§ОЭЖдгІзХЕчзгЕФвЛжжФмСПзДЬЌЃЌМШШЛЕчзгЕФФмСПзДЬЌЪЧВЛСЌајЕФЃЌвђДЫСПзгЪ§ЕФжЕвВЪЧВЛСЌајЕФЁЃ
БэЪОЙьЕРдЫЖЏЕФШ§ИіСПзгЪ§ЕФзщКЯЖдгІзХвЛИідзгЙьЕРЃЈГЦЮЊЕчзгЕФвЛИіСПзгЬЌЃЉЃЌгЩгкетШ§ИіСПзгЪ§жЎМфДцдкзХЬиЖЈЕФжЦдМЙиЯЕЃЌЪЙжїСПзгЪ§nШЗЖЈЕФУПвЛИіЕчзгВужаЃЌдзгЙьЕРжжРрКЭИіЪ§ЖМЪЧУїШЗЕФЁЃЫфШЛЕЅЕчзгЬхЯЕжаЃЌдзгКЫЭтжЛгавЛИіЕчзгЃЌЕЋетаЉВЛЭЌЕчзгВуКЭВЛЭЌдзгЙьЕРЕФДцдкЃЌБэУїдзгКЫЭтЕФвЛИіЕчзггаПЩФмГіЯжЕФВЛЭЌФмСПзДЬЌЃЈМЄЗЂЬЌЃЉЁЃ
ЫФИіСПзгЪ§ЙцЖЈСЫКЫЭтЕчзгЕФдЫЖЏзДЬЌЃЌУПИіЕчзгЖМПЩвдгУЩЯЪіЕФЫФИіСПзгЪ§ЕФвЛЬзЪ§ОнРДУшЪіЦфдЫЖЏзДЬЌЃЈЖдгІзХвЛИіВЈКЏЪ§ЃЉЃЌЭЌвЛдзгжаУЛгаЫФИіСПзгЪ§ЭъШЋЯрЭЌЕФЕчзгЁЃЛЛОфЛАЫЕЃЌдкЭЌвЛдзгжаЕФИїИіЕчзгЃЌЫќУЧЕФдЫЖЏзДЬЌВЛПЩФмЭъШЋЯрЭЌЃЌМДЫФИіСПзгЪ§жажСЩйгавЛИіСПзгЪ§ЪЧВЛЭЌЕФЁЃЊЅ
ЖдЕЅЕчзгЬхЯЕЃЌПЩгУЕЅЕчзгЕФбІЖЈкЬЗНГЬДІРэЃЌжЛашДІРэ1ИіКЫКЭ1ИіЕчЕФЮќв§зїгУЁЃЖдЖрЕчзгЬхЯЕЃЌвЊгУЖрЕчзгЕФгУбІЖЈкЬЗНГЬДІРэЃЌNИіЕчзггы1ИіКЫЕФЮќв§зїгУЃЌвЊДІРэNИіЕчзгжЎМфЕФХХГтзїгУЁЃЖдгкетжжИДдгЙиЯЕЃЌПЩвдНќЫЦЕигУЦСБЮаЇгІКЭзъДЉаЇгІЕШаЇДІРэЁЃ
КЌгаNИіЕчзгЕФдзгжаЃЌЕчзгiГ§СЫЪмЕНдзгКЫ(КЫЕчКЩЮЊZ)ЕФЮќв§ЭтЃЌЭЌЪБЛЙЪмЕНЦфЫћЃЈNЃЃБЃЉИіЕчзгЕФХХГтЁЃЪЕМЪЩЯЯрЕБгкВПЗжЕиЕжЯћСЫКЫЕчКЩZЖдЕчзгЃщЕФЮќв§ЃЌДгЖјв§ШыгааЇКЫЕчКЩZ*ЕФИХФюЃКЃк*ЊЌЃНЃкЃІв
Ів--ЦСБЮГЃЪ§ЃЌЕШгкNЃЃБИіЕчзгЖдЕчзгiЕФЦСБЮзїгУЕФзмКЭЕЅЕчзгЬхЯЕЃЌЕчзгЃЈЙьЕРЃЉЕФФмСПНігыжїСПзгЪ§nгаЙиЃКЃХЃюЃНЃЃвЁЄ(ZЃВ/ЃюЃВ)
ЖдЖрЕчзгЬхЯЕЃЌЕчзгЃЈЙьЕРЃЉЕФФмСПЛЙгыНЧСПзгЪ§lгаЙиЃКЃХЃюЃЌЃьЃНЃЃвЁЄ(Z*ЃВ/ЃюЃВ)
ЦфжаЃКЃвЃНЃБЃГ.ЃЖevЛђЃвЃН2.179ЁСЃБЃАЃЃБЃИЃЪ
гЩЙЋЪНВЛФбПДГіЃЌЖдЯрЭЌдзгЃЈКЫЕчКЩZвЛЖЈЃЉЃЌЕБЖдЕчзгiЕФЦСБЮГЃЪ§ІвдНДѓЃЌЖдЫќЕФгааЇКЫЕчКЩZ*дНаЁЃЌЕчзгiЕФФмСПОЭдНИпЃЌЫљвдЦСБЮаЇгІНсЙћЪЙЕчзгЕФФмСПЩЯЩ§ЁЃ
ЃюЯрЭЌЃЈЭЌВуЃЉЖјЃьВЛЭЌЕФЙьЕРЃЌЦфЕчзгдЦЕФОЖЯђЗжВМЧщПіВЛЭЌЃЌЃьдНаЁЕФЙьЕРЦфЗхЪ§дНЖрЃЈЗхЪ§=n-lЃЉЃЌВПЗжЕчзгдЦЕчзгДЉЙ§ФкВуЖјИќППКЫИќНќЃЌетжжЯжЯѓГЦЮЊзъДЉаЇгІЁЃЕчзгЭЈЙ§зъДЉаЇгІФмЙЛЛиБмЦфЫћЕчзгЖдЫќЕФЦСБЮзїгУЃЌЫљИаЪмЕНЕФгааЇКЫЕчКЩЃк*жЕдНИпЃЌЪЙФмСПНЕЕЭЕЭЁЃ
УРЙњжјУћНсЙЙЛЏбЇМвБЋСжИљОнДѓСПЙтЦзЪЕбщЪ§ОнМАРэТлМЦЫуЕФНсЙћЃЌЬсГіСЫЖрЕчзгдзгжаЙьЕРЕФНќЫЦФмМЖЭМЁЃетИіЭМЕФФмМЖЫГађгыЭтВуЙьЕРЕчзгЕФЬюГфЫГађЪЧвЛжТЕФЃЌвђДЫНќЫЦФмМЖЭМЪЕМЪЩЯЪЧЕчзгЬюГфЫГађЭМЁЃЊЄ
КЫЭтЕчзгЕФХХВМЙцТЩЁЊЁЊХнРћ(Pauli)ВЛЯрШндРэЃКЁАдкЭЌвЛдзгжаУЛгаЫФИіСПзгЪ§ЭъШЋЯрЭЌЕФЕчзгЁБЛђепЃКЁАЭЌвЛдзгЙьЕРжЛФмШнФЩСНИізда§ЯрЗДЕФЕчзгЁБУПвЛЕчзгВуЁЂУПвЛЕчзгбЧВуЫљФмШнФЩЕФЕчзгЪ§ЖМЪЧвЛЖЈЕФЁЃ
зюЕЭФмСПдРэЃКЁАдкВЛЮЅБГХнРћдРэЕФЧАЬсЯТЃЌКЫЭтЕчзгдкИїдзгЙьЕРжаЕФХХВМЗНЪНгІЪЙећИідзгЕФФмСПДІгкзюЕЭЕФзДЬЌЁЃЁБгІЕБАДееНќЫЦФмМЖЭМЕФЫГађЬюГфЕчзгЁЃ
КщЬиЙцдђЃКдкФмСПЯрЭЌЕФЙьЕРЩЯХХВМЕчзгЪБЃЌзмЪЧвдзда§ЯрЭЌЕФЗНЯђгХЯШЗжеМВЛЭЌЕФЙьЕРЃЌетбљЬхЯЕЕФФмСПНЯЕЭЁЃвђДЫЃЌдквЛИібЧВужаЃЌАыГфТњзДЬЌЪЧИУбЧВужазда§ГЩЕЅзюЖрЕФзДЬЌ(Ѓ№3ЃЌЃф5ЃЌЃц7)ЃЌФмСПзюЕЭЁЃДЫЭтЃЌбЧВуДІгкШЋГфТњ(ШчЃ№6ЃЌЃф10ЃЌЃц14)зДЬЌЪБвВЪЧвЛжжЕЭФмСПЕФзДЬЌЁЃ
жмЦкЃК7ИіФмМЖзщЕФДцдкЪЧдЊЫиБэБЛЛЎЗжГЩ7ИіжмЦкЕФБОжЪдвђЁЃ
жмЦкКХЪ§=ФмМЖзщКХЪ§=ЕчзгВуЪ§=зюДѓжїСПзгЪ§Ѓюmax
УПжмЦкЕФдЊЫиИіЪ§=ИУжмЦкдЊЫиЭтВуЙьЕРЫљФмШнФЩЕФЕчзгЪ§ЁЃ
УПвЛжмЦкДгЃюЃѓбЧВуПЊЪМЃЌЕНЃюЃ№бЧВуНс(ЕквЛжмЦкжЛгаЃБЃѓбЧВу)ЁЃ
зхЃКУПжмЦкдЊЫиЕФЕчзгВуНсЙЙГіЯжжмЦкадЕФБфЛЏЃЌНЋЯрЭЌЕФМлЕчзгВуНсЙЙЕФдЊЫиЙщЮЊЭЌвЛСаЃЈЙВЗж18СаЃЉЃЌетЪЧжмЦкБэЗжзхЕФвРОнЁЃ
жїзхеМ8СаЃЌЗж8зхЃЌЗВЪЧзюКѓвЛИіЕчзгЬюШыЃюЃѓЛђЃюЃ№ФмМЖЕФдЊЫиГЦЮЊжїзхдЊЫиЁЃ
жїзхдЊЫиЕФМлЕчзгХХВМЭЈЪНЃКns1-2np0-6
ИБзхеМ10СаЃЌЗж8зхЃЈЕк8зхеМ3СаЃЉЃЌЗВЪЧзюКѓвЛИіЕчзгЬюдкЃЈn-1ЃЉЃфФмМЖЛђ(n-2)ЃцФмМЖЩЯЕФдЊЫиЃЌГЦЮЊИБзхдЊЫиЁЃ
ИБзхдЊЫиЕФМлЕчзгХХВМЭЈЪНЃК(n-2)f1-14(n-1)d1-10ns1-2
ЗжЧјЃКНЋНсЙЙЯрЫЦЕФзхЛЎЮЊЭЌвЛЧј
ЃѓЧјЃКЗВЪЧМлЕчзгВузюИпФмМЖЮЊЃюЃѓ1-2ЕчзгзщЬЌЕФдЊЫиЃЌГЦЃѓЧјдЊЫиЁЃЫќАќРЈЂёЃСКЭЂђAСНИіжїзхдЊЫиЁЃ
pЧјЃКЗВЪЧМлЕчзгВуЩЯОпгаns2npЃБ-6ЕчзгзщЬЌЕФдЊЫиГЦЮЊpЧјдЊЫиЁЃЫќАќРЈЂѓA-ЂїAвдМА0зхЕФжїзхдЊЫиЁЃ
ЃфЧјЃКМлЕчзгВуЩЯОпга(n-1)dЃБ-ЃЙnsЃБ-ЃВЕчзгзщЬЌЕФдЊЫиГЦЮЊЃфЧјдЊЫиЁЃЫќАќРЈЂѓЃТ-Ђј6ИіИБзхдЊЫиЁЃ
ЃфЃѓЧјЃКМлЕчзгВужаОпга(n-1)dЃБЃАnsЃБ-ЃВЕчзгзщЬЌЕФдЊЫиГЦЮЊЃфЃѓЧјдЊЫиЁЃЫќАќРЈЂёЃТЁЂЂђЃТСНИіИБзхдЊЫиЁЃ
ЃцЧјЃКМлЕчзгВужаОпга(n-2)fЃБ-ЃБЃД(n-1)dЃБnsЃВЕчзгзщЬЌЕФдЊЫиГЦЃцЧјдЊЫи,ячЯЕдЊЫиКЭяЙЯЕдЊЫиЪєгкЃцЧјдЊЫиЁЃЊІ
дЊЫиЕФаджЪЪЧЦфдзгНсЙЙЕФЗДгГЃЌЫцзХдзгађЪ§ЕФдіДѓЃЌдзгЕФЕчзгВуНсЙЙГЪжмЦкадЕФБфЛЏЃЌвђДЫгыЕчзгВуНсЙЙгаЙиЕФдЊЫиЕФЛљБОаджЪЃЌвВГЪЯжжмЦкадБфЛЏЁЃ
ЙТСЂдзгЕФАыОЖЃКдзгЕФЛљЕчзгзщЬЌжаЃЌеМОнзюИпФмМЖЩЯЕФЕчзгЕНдзгКЫЕФОрРыЁЃгЩгкЕчзгЕФВЈЖЏадКЭВЛШЗЖЈЙиЯЕЖјВтВЛзМЁЃПЩвдгУЛљЬЌдзгзюЭтВуЕФдзгЙьЕРЕФАыОЖНќЫЦДњБэЦфАыОЖЙТСЂдзгЕФАыОЖЁЃЃђЃю=ЃНЃю2Ѓс0/ЃЈЃк*ЃЉЪЕМЪдзгЕФАыОЖЃКЦфДѓаЁгыЦфЫљДІЛЗОГгаЙиЃЌШЁОігкЫќгыЛЗОГжадзгжЎМфзїгУСІЕФаджЪЃЌЗжЮЊЃК
(a)дзгЕФЙВМлАыОЖЈC-ЭЌжждЊЫиЕФСНИідзгAЃЌвдЙВМлМќНсКЯГЩЗжзгAЃВЪБЃЌСНдзгЕФКЫМфОрЕФвЛАыЃЌЮЊAдзгЕФЙВМлАыОЖЁЃ
(b)ЗЖЕТЛЊАыОЖЈCдквдЗЖЕТЛЊСІаЮГЩЕФЗжзгОЇЬхжаЃЌВЛЪєгкЭЌвЛИіЗжзгЕФСНИізюНгНќдзгЕФКЫМфОрЕФвЛАыЁЃ
(c)дзгЕФН№ЪєАыОЖ-дкН№ЪєОЇЬхжаЃЌСНЯрЛЅНгДЅдзгЕФКЫМфОр(Ры)ЕФвЛАыЁЃ
дзгАыОЖЕФЯрЖдДѓаЁЃКЗЖЕТЛЊАыОЖЃОздгЩдзгЁААыОЖЁБЃОН№ЪєАыОЖЃОЙВМлАыОЖ
дзгАыОЖЕФБфЛЏЙцТЩЃК
ЭЌжмЦкБШНЯЃКжїСПзгЪ§ЃюЯрЭЌЃЌгааЇКЫЕчКЩЪ§Ѓк*дНДѓЃЌЖдзюЭтбЧВужаЕчзгЕФЮќв§СІОЭдНДѓЃЌЯргІЕФдзгАыОЖдНаЁЃЛ
жїзхдЊЫиЃКДгзѓЕНгвЁњдзгАыОЖЁ§ЃЈЕЋЖшаддЊЫиАыОЖЭЛШЛдіДѓЃЉ
ИБзхдЊЫиЃКДгзѓЕНгвЁњдзгАыОЖЁ§ЃЈЕЋЕНСЫЂёBКЭЂђBСНИіИБзхдЊЫидзгАыОЖУїЯдЕидіДѓСЫЃЉ
жїзхдЊЫидзгАыОЖЕнБфЙцТЩУїЯдЃЌвђЮЊгааЇКЫЕчКЩЃк*ЕндіУїЯдЁЃ
ЭЌзхБШНЯЃКгааЇКЫЕчКЩЪ§Ѓк*ЛљБОЯрЭЌЃЌзюЭтбЧВуЕФжїСПзгЪ§ЃюдНДѓЃЌзюЭтбЧВуЕчзгЕФФмСПдНДѓЃЌвђЖјЫќУЧРыКЫЕФОрРывВдНДѓЃЌМДдзгАыОЖдНДѓЁЃЊЅ
жїзхдЊЫиЃКздЩЯЖјЯТдзгАыОЖдіДѓЁЃ
ИБзхдЊЫиЃКrЃЈЫФжмЦкЃЉЃМrЃЈЮхжмЦкЃЉЁжrЃЈСљжмЦкЃЉЃЌгЩячЯЕЪеЫѕв§Ц№
ЕчРыФмЃКвЛИіДІгкЛљЬЌЕФЦјЬЌздгЩдзгЪЇШЅвЛИіЕчзгЃЌБфГЩе§вЛМлЕФЦјЬЌе§РызгЫљашвЊЮќЪеЕФзюаЁФмСПжЕЃЌБЛЖЈвхЮЊИУдзгЕФЕквЛЕчРыФмЃЩ1ЃЛШчдйЪЇШЅвЛИіЕчзгЃЌБфГЩе§ЖўМлЕФЦјЬЌе§РызгЫљашвЊЮќЪеЕФзюаЁФмСПжЕЃЌБЛЖЈвхЮЊЕкЖўЕчРыФмI2ЃЛЦфгрРрЭЦЁЃ
ЕчРыФмБфЛЏЙцТЩЃК
ЭЌжмЦкБШНЯЃКжїСПзгЪ§ЯрЭЌЃЌгааЇКЫЕчКЩЪ§дНДѓЃЌЕчРыФмдНДѓЁЃ
жїзхдЊЫиЃКзѓ-гвЃКЕквЛЕчРыФмвРДЮУїЯддіДѓЃЈЕЋЦфжагааЉЧњелЃЌВЛЙцдђЕФдвђЃКЖрЪ§гыШЋПеЁЂШЋТњКЭАыТњЙЙаЭЪЧБШНЯЮШЖЈЕФЙЙаЭгаЙиЁЃЃЉЁЃ
ИБзхдЊЫиЃКзѓ-гвЃКЦфЕквЛЕчРыФмВЛЙцдђЕидіМгЃЌЧвдіМгЕФЗљЖШКмаЁЁЃ
ЭЌзхБШНЯЃКгааЇКЫЕчКЩЪ§ЯрЭЌЃЌжїСПзгЪ§дНДѓЃЌЕчРыФмдНаЁЁЃ
жїзхдЊЫиЃКздЩЯЖјЯТЃЌЫцжїСПзгЪ§ЕндіЖјУїЯдЕивРДЮМѕаЁЁЃ
ИБзхдЊЫиЃКздЩЯЖјЯТЃЌБфЛЏЗљЖШНЯаЁЃЌЧвЙцТЩадвВВюЁЃЃЈЕкСљжмЦкЕФИБзхдЊЫиЕФЕквЛЕчРыФмЗДЖјБШЕкЫФЁЂЮхжмЦкЕФДѓЃЌжївЊЪЧгЩЁАячЯЕЪеЫѕЁБаЇгІв§Ц№ЕФЃЉЁЃЊЅ
ЕчзгЧзКЭФмЃКвЛИіДІгкЛљЬЌЕФЦјЬЌздгЩдзгЛёЕУвЛИіЕчзгБфГЩИКвЛМлЦјЬЌздгЩРызгЪБЫљЮќЪе(е§жЕ)ЛђЗХГі(ИКжЕ)ЕФзюаЁФмСПЃЌГЦИУдзгЕФЕквЛЕчзгЧзКЭФмЁЃЕчзгЧзКЭФмвВгаЕквЛЁЂЖўМЖЕШЕчзгЧзКЭФмжЎЗжЃЌЗжБ№БэЪОЮЊY1ЁЂY2ЁЃ
ЕчзгЧзКЭФмЕФБфЛЏЙцТЩЃКЕчзгЧзКЭФмвЛАуЫцдзгАыОЖМѕаЁЖјдіДѓЁЃетЪЧвђЮЊдзгАыОЖМѕаЁЃЌКЫЕчКЩЖдЕчзгЕФЮќв§СІОЭдіЧПЃЌдзгдђвзНсКЯЭтРДЕчзгЖјЗХГіФмСПЁЃ
жїзхдЊЫиЃК
ЭЌвЛжмЦкЃЌзѓ-гвЃЌдзгАыОЖМѕЩйЃЌЕчзгЧзКЭФмж№НЅдіДѓЁЃ
ЭЌвЛжїзхЃЌдзгАыОЖздЩЯЖјЯТдіДѓЃЌЕчзгЧзКЭФмздЩЯЖјЯТМѕаЁЁЃЃЈЕЋЪЧУПвЛзхПЊЭЗдЊЫиЕФЕчзгЧзКЭФмЗДЖјаЁаЉЃЌетЪЧвђЮЊЧАепЕФдзгАыОЖБШКѓепЕФаЁЕУЖрЃЌЕчзгМфГтСІЧПЃЉ
ИБзхдЊЫиЃКЖдгкИБзхдЊЫиЃЌЫќУЧЕФЕчзгЧзКЭФмЪ§ОнБШНЯЩйЃЌБфЛЏЙцТЩадвВВЛУїЯдЁЃЊЅ
дЊЫиЕчИКадЕФИХФюЃКЫљЮНдЊЫиЕчИКадЪЧжИдзгдкЗжзгжаЮќв§ГЩМќЕчзгЕФФмСІЃЌГЃгУІжБэЪОЁЃдЊЫиЕчИКаддНДѓЃЌИУдЊЫидзгдкЗжзгжаЮќв§ГЩМќЕчзгЕФФмСІдНДѓЃЌЗДжЎдНаЁЁЃ
ЕчИКадБфЛЏЙцТЩЃКетКЭгааЇКЫЕчКЩКЭдзгАыОЖЕФБфЛЏЙцТЩЪЧвЛжТЕФЁЃ
жїзхдЊЫиЃКЭЌвЛжмЦкздзѓжСгвЕчИКаддіДѓЃЛЭЌвЛзхздЩЯЖјЯТЕчИКадМѕаЁЃЌ
ИБзхдЊЫиЃКЭЌвЛжмЦкздзѓжСгвЕчИКадТдгадіМгЃЛЭЌвЛзхжаЕФЙцТЩЃК
ЊЉІж(ЕквЛЙ§ЖЩЯЕ)ЃОІж(ЕкЖўЙ§ЖЩЯЕ)ЁжІж(ЕкШ§Й§ЖЩЯЕ)
дкжмЦкБэжаЃЌгвЩЯЗНІж(ЃЦ)зюДѓЃЈ3.98ЃЉЃЌзѓЯТЗНІж(ЃУЃѓ)зюаЁЃЈ0.79ЃЉЃЛ
ЕчИКадЕФгІгУЃКa.ХаЖЯдЊЫиЕФН№ЪєадКЭЗЧН№ЪєадЃЛЕчИКаддНДѓЃЌН№ЪєаддНШѕЃЌЗЧН№ЪєаддНЧПЃЛЕчИКаддНаЁЃЌН№ЪєаддНЧПЃЌЗЧН№ЪєаддНШѕЁЃ
b.ХаЖЯЛЏбЇМќЕФаджЪКЭМќЕФМЋадЃЛІЄXЃМ1.7МЋадЙВМлМќЃЌІЄXЃО1.7РызгМќ
аЁНсЃЌдкБОеТжаЃЌЮвУЧЬжТлСЫдзгЕФНсЙЙЃЌжиЕуеЦЮеЫФИіСПзгЪ§ЕФвтвхвдМАЫќУЧЕФШЁжЕЙцТЩМАдзгаджЪЕФжмЦкадЁЃЧыЭЌбЇУЧНсКЯПЮКѓЯАЬтЃЌНјвЛВНРэНтдзгНсЙЙМАЦфжмЦкадБфЛЏЙцТЩЁЃ
ЕкСљеТ ЗжзгЕФНсЙЙгыаджЪ
ЮвУЧЖМжЊЕРЃЌЗжзгЪЧВЮгыЛЏбЇЗДгІЕФЛљБОЕЅдЊжЎвЛЁЃвВЪЧБЃГжЮяжЪЛЏбЇаджЪЕФзюаЁЮЂСЃЁЃвђДЫЃЌЖдЗжзгФкдзгМфЕФГЩМќЧщПіНјааЗжЮіЃЌНјЖјЬжТлЗжзгЕФаЮГЩЙ§ГЬЃЌФмЙЛАяжњЮвУЧИќКУЕФРэНтЗжзгЕФНсЙЙгыаджЪжЎМфЕФЙиЯЕЁЃдкБОеТжаЃЌЮвУЧНЋдкЕкАЫеТ дзгНсЙЙЕФЛљДЁЩЯЃЌжиЕуЬжТлЙВМлМќРэТлЃЌКЭЗжзгЙЙаЭЕШЮЪЬтЁЃЮвУЧЖМжЊЕРЃЌЛЏбЇМќЪЧжИЕФдзгЛђРызгжЎМфЕФЧПЯрЛЅзїгУЃЌжївЊАќРЈРызгМќЃЌЙВМлМќЃЌН№ЪєМќШ§жжРраЭЁЃдкБОеТжаЃЌЮвУЧНЋжиЕуЬжТлЙВМлМќРэТлЃЌАќРЈЃЌ1916ФъLewisРэТлЃЌМлМќРэТл(1927ФъЃЌ1930Фъ)ЃЌвдМАдкДЫЛљДЁЩЯЗЂеЙЕФМлВуЕчзгЖдЛЅГтРэТл(1940Фъ)КЭдгЛЏЙьЕРРэТл(1931Фъ)ЁЃДЫЭтЃЌЮвУЧЛЙвЊбЇЯАЃЌНЈСЂдкдзгЙьЕРРэТлЛљДЁЩЯЕФЗжзгЙьЕРРэТл(20ЪРМЭ20ФъДњФЉ)ЁЃМлМќРэТлКЭЗжзгЙьЕРРэТлИїгаЬиЩЋЃЌПЩвдАяжњЮвУЧдЄВтЃЌЗжЮіЃЌНтОіЗжзгЕФНсЙЙКЭаджЪЃЌЫќУЧЙВЭЌЙЙГЩСЫЙВМлМќРэТлЁЃ
ЙВМлМќЕФИХФюЪЧУРЙњШЫТЗвзЫЙЃЈG.N.LewisЃЉгк1916ФъЪзЯШЬсГіЕФЃЌТЗвзЫЙШЯЮЊЃЌЗжзгжаУПИідзгЖМОпгааЮГЩРрЫЦгкЖшаддзгЕФЮШЖЈЕчзгНсЙЙЕФЧуЯђЃЌВЂЭЈЙ§дзгМфЁАЙВгУЁБЕчзгЖдЕФЗНЪННсКЯГЩЗжзгЁЃгЩДЫаЮГЩЕФЛЏбЇМќГЦЮЊЙВМлМќ(covalent bond)ЃЌетжжЗжзгГЦЮЊЙВМлЗжзгЁЃДЫКѓдкСПзгСІбЇбаОПH2ЗжзгЙВМлМќБОжЪЕФЛљДЁЩЯЗЂеЙЦ№СЫЙВМлМќЕФМлМќРэТлЃЈvalence bond theory, VBTЃЉЃЌгжГЦЕчзгХфЖдЗЈ(electron pairing method)ЁЃЫќЮЊЙВМлМќЕФБОжЪдкгкЙВЯэЕчзгЖдЕФЙлЕуЬсЙЉСЫРэТлЛљДЁЁЃ
МлМќРэТлШЯЮЊЃЌвЛИіЙВМлМќЕФаЮГЩБиаыОпБИвдЯТСНИіЬѕМўЃЈвЊЕуЃЉЃК
ЕчзгХфЖдГЩМќдРэЃКВЮгыаЮГЩвЛИіЙВМлМќЕФСНИіЃЈЛљЬЌЃЉдзгЃЌИїздЖМБиаыЬсЙЉвЛИізда§(spin)УЛгаХфЖдЕФЕчзгЃЌетСНИіЕчзгдкзда§ЯрЗДЕФЬѕМўЯТЃЌВХФмЙЛХфЖдаЮГЩвЛИіЙВМлМќЁЃШчЙћдзгжаУЛгаЮДХфЖдЕчзгЃЌвЛАуВЛаЮГЩЙВМлМќЃЌЖјвбХфЖдГЩМќЕФЕчзгВЛФмдйВЮгыаЮГЩаТЕФЙВМлМќЁЃ
ЭЈЙ§H2ЗжзгЕФФмСПЧњЯпЭМЃЌПЩвдЫЕУїHдзгаЮГЩЙВМлМќЕФЬѕМўЁЃШчЙћСНИіHдзгЕФГЩЕЅЕчзгзда§ЗНЯђЯрЗДЃЌдђСНИідзг1sЙьЕРЛЅЯржиЕўЃЌСНКЫМфЕФЕчзгдЦУмЖШдіДѓЃЌЬхЯЕЕФФмСПНЕЕЭЁЃдкСНИідзгКЫжЎМфЙЙГЩвЛИіИКЕчКЩЕФЁАЧХЁБЃЌАбСНИіДје§ЕчКЩЕФКЫЮќв§дквЛЦ№ЁЃЕБКЫМфОрДяЕНЦНКтОрРыR0ЪБЃЌЬхЯЕЕФФмРэДяЕНзюЕЭЕуЁЃШєСНИідзгНјвЛВНППНќЃЌгЩгкСНКЫМфЕФПтТиХХГтСІж№НЅдіДѓЖјЪЙЬхЯЕЕФФмСПЩ§ИпЁЃвђДЫЃЌСНИіHдзгдкЦНКтОрРыR0ДІПЩвдаЮГЩЮШЖЈЕФH2ЗжзгЁЃЪЕбщВтЕУR0=74pmЃЌФмСПНЕЕЭЕФЪ§жЕ![]() ЃЌЫќНќЫЦЕШгкH2ЗжзгЕФМќФмЁЃ
ЃЌЫќНќЫЦЕШгкH2ЗжзгЕФМќФмЁЃ
ШчЙћСНИідзгЕФЕчзгзда§ЗНЯђЯрЭЌЃЌдђСНКЫМфЕФЕчзгдЦУмЖШЯЁЪшЃЌЕчзгЛЅЯрХХГтЁЃЬхЯЕЕФФмРэИпгкЕЅЖРДцдкЕФHдзгФмСПжЎКЭЃЌетОЭвтЮЖзХЫќУЧВЛПЩФмНсКЯГЩH2ЗжзгЁЃ
ПЩМћЃЌЙВМлМќЕФаЮГЩЪЧгЩгкСНдзгЯрЛЅППНќЪБЃЌОпгазда§ЗНЯђЯрЗДЕФЮДГЩЖдЕчзгЕФЯргІдзгЙьЕРЛЅЯржиЕўЃЌЕчзгдЦУмМЏдкСНИідзгКЫжЎМфЃЌДгЖјЪЙЬхЯЕЕФФмСПНЕЕЭЁЃ
ЕЋЪЧЃЌвЛИідзгжаЕФЮДХфЖдЕчзгЪ§ЕФИХФювВВЛЪЧвЛГЩВЛБфЕФЁЃзїЮЊЖдМлМќРэТлЕФживЊВЙГфКЭЗЂеЙЃЌгаШЫЬсГіМЄЗЂГЩМќЕФЙлЕуЁЃЫљЮНМЄЗЂГЩМќЃЌЪЧжИВЮгыГЩМќЕФдзгЃЌЦфМлЙьЕРЩЯвбХфЖдЕФЕчзгЃЌПЩФмБЛМЄЗЂЕНЯрСкЕФПеМлЙьЕРЩЯЃЌВ№ЗжГЩСНИіЮДХфЖдЕчзгЁЃМЄЗЂЕчзгЫљашвЊЕФФмСПЃЌПЩгЩаЮГЩЙВМлМќЪБЫљЗХГіЕФФмСПгшвдВЙГЅЁЃР§ШчЃЌBeдзгЕФЛљЕчзгзщЬЌЮЊ1s22s2ЃЌВЂЮоЮДГЩЖдЕчзгЁЃЦфМлЕчзгВуЕФЛљЕчзгзщЬЌ2s22p0ЃЌБЛМќКЯдзгClМЄЗЂГЩ2s12p1ЃЌЪЙBeдзгВњЩњСНИіЮДХфЖдЕчзгЃЌПЩаЮГЩСНИіЙВМлМќЃЌвђДЫВХгаBeCl2етРрЗжзгЕФДцдкЁЃ
дзгЕФМлЕчзгВугаЖрЩйЮДХфЖдЕФЕчзгЃЌаЮГЩЗжзгЪБЃЌОЭЛсаЮГЩЖрЩйИіЙВМлМќЁЃР§ШчH3CЁЊCH3ЮЊЙВМлЕЅМќЁЂCH2=CH2ЮЊЙВМлЫЋМќ(doublebond)ЁЂHC=CHЮЊЙВМлВЮМќ(triplebond)ЁЃЕчзгХфЖдГЩМќдРэОіЖЈСЫЙВМлМќОпгаБЅКЭадЁЃ
ЙьЕРзюДѓжиЕўдРэЃКдкТњзуЕквЛИіЬѕМўЕФЧАЬсЯТЃЌжЛгаСНзда§ЯрЗДЕчзгЕФЕчзгдЦжиЕўВХФмаЮГЩвЛИіЙВМлМќЃЌВЂЧвЕчзгдЦжиЕўЧјгђдНДѓЃЌЬхЯЕФмСПНЕЕУдНЕЭЃЌаЮГЩЕФЙВМлМќВХдНЮШЖЈЁЃетОЭЪЧЕчзгдЦзюДѓжиЕўдРэЁЃ
Г§sЕчзгдЦЪЧЧђаЮЖдГЦЕФвдЭтЃЌЦфгрдзгЙьЕРЕФЕчзгдЦЖМОпгаШЗЖЈЕФМЋжЕШЁЯђЁЃШчЙћsЕчзгдЦгыpxЕчзгдЦжиЕўЃЌдђБиаыбиЖдгІЕФxжсЕФЗНЯђжиЕўВХЪЧзюДѓжиЕўЁЃЙьЕРзюДѓжиЕўдРэОіЖЈСЫЙВМлМќОпгаЗНЯђадЁЃИљОнЕчзгдЦжиЕўГЬЖШВЛЭЌЃЌПЩЖдЙВМлМќНјааЗжРрЁЃ
ЙВМлМќЕФЬиЕуЃК
ЃЈ1ЃЉЙВМлМќОпгаЗНЯђад
ЙВМлМќЕФЗНЯђадЪЧгЩЙьЕРзюДѓжиЕўдРэОіЖЈЕФЃЌвђЮЊвЊЪЕЯжЙьЕРЕФзюДѓжиЕўЃЌБиаыдкКЫМфОрВЛБфЕФЧщПіЯТЃЌСНЙьЕРбиЦфМЋжЕзюДѓЕФЗНЯђжиЕўВХЪЧзюДѓЕФжиЕўЁЃгЩгкЙВМлМќдкЗжзгжаЕФЯрЖдШЁЯђЪЧШЗЖЈЕФЃЌФЧУДгЩЙВМлМќСЌНгЕФдзгдкЗжзгжаЕФЯрЖдЮЛжУвВЪЧШЗЖЈЕФЃЌМДЗжзгЕФМИКЮЙЙаЭЪЧШЗЖЈЕФЁЃдкЖрдзгЗжзгжаЃЌЙВМлМќЕФЗНЯђадгазХживЊзїгУЃЌЫќВЛНіОіЖЈСЫЗжзгЕФМИКЮЙЙаЭЃЌЖјЧвЛЙгАЯьзХЗжзгЕФМЋадКЭЖдГЦадЕШЗжзгаджЪЁЃ
ЃЈ2ЃЉЙВМлМќОпгаБЅКЭад
ЙВМлМќЕФБЅКЭадЪЧЕчзгХфЖдГЩМќдРэОіЖЈЕФЃЌгЩгкУПжждЊЫиЕФдзгЫљФмЬсЙЉЕФМлЙьЕРЪ§КЭаЮГЩЗжзгЪБЫљашЬсЙЉЕФЮДГЩЕчзгЪ§ЪЧвЛЖЈЕФЃЌЫљвджЛвЊФмШЗЖЈФГИідзгЕФЮДХфЖдЕчзгЪ§ЃЈАќРЈМЄЗЂКѓаЮГЩЕФЮДХфЖдЕчзгЪ§ЃЉЃЌОЭФмЭЦЫуГіИУдзгПЩФмаЮГЩЕФзюЖрЙВМлМќЪ§ЁЃ
ЙВМлМќЕФРраЭЃКЙВМлМќЕФаЮГЩЪЧСНЮДХфЖдЕчзгЕФЕчзгШЅЯрЛЅжиЕўЕФНсЙћЁЃИљОнЕчзгдЦЕФжиЕўЗНЯђЃЌЗНЪНМАжиЕўКѓЕчзгдЦЗжВМЕФЖдГЦадЃЌПЩНЋГЃМћЕФЙВМлМќЗжЮЊСНРрЃЌМДІвМќКЭІаМќСНРрЁЃИљОнЙВМлМќжаЙВгУЕчзгЖдЕФРДдДВЛЭЌЃЌгжПЩвдНЋЙВМлМќЗжЮЊЦеЭЈЙВМлМќКЭХфЮЛЙВМлМќЃЈcomplex valence bondЃЉСНРрЁЃ
ІвМќ(Ів bond)ШчЙћИїВЮгыГЩМќЕФЕчзгдЦЃЌбиМќжсЃЈМДГЩМќСНдзгКЫжЎМфЕФСЌЯпЃЉЗНЯђЕФЗжВМЪЧдВжљаЮЖдГЦЕФЃЌЫќУЧжиЕўжЎКѓЕФЕчзгдЦШдБЃГждРДЕФдВжљаЮЖдГЦадЃЌЫљаЮГЩЙВМлМќГЦЮЊІвМќЁЃ
Р§ШчдкCl2ЗжзгжаЃЌУПИіClдзгЕФ3pxЙьЕРЖМгавЛИіЮДХфЖдЕчзгЃЌетСНИі3pxЕчзгдЦЖдxЃЈМќЃЉжсГЪдВжљаЮЖдГЦЃЌЖўепбиxжсЗНЯђжиЕўКѓЕФЕчзгдЦЗжВМЖдxжсШдОпгадВжљаЮЖдГЦадЃЈМћЭМ9Ѓ3ЃЉЃЌЫљвдЕчзгдЦpx-pxбиxжсжиЕўаЮГЩЕФБиШЛЪЧІвМќЁЃЦфЫћШчs-sЕчзгдЦЕФжиЕўЃЌs-pЕчзгдЦЕФжиЕўЕШЖМаЮГЩІвМќЁЃ
ІаМќ(Іа bond)ШчЙћИїВЮгыГЩМќЕФЕчзгдЦЃЌЖдЙ§МќжсЕФЦНУцГЪЖдГЦЗжВМЃЌЖјжиЕўКѓЕФЕчзгдЦШдШЛБЃСєетжжЖдГЦадЃЌетбљаЮГЩЕФЙВМлМќГЦЮЊІаМќЁЃР§ШчЃЌдкN2ЗжзгжаЃЈЭМ2Ѓ4ЃЉЃЌNдзгЕФГЩМќЙьЕРМАГЩМќЕчзг![]() ,ЃЌЕБЫќУЧбиМќжсЗНЯђЯрЛЅППНќЪБЃЌ2px-2pxжиЕўГЪдВжљаЮЖдГЦЃЌаЮГЩвЛИіІвМќЃЛ2pz-2pzЙьЕРбиМќжсЗНЯђжиЕўЪБЃЌдђгавЛИіЖдГЦУцЁЃетИіЖдГЦУцЭЈЙ§МќжсЖјДЙжБгкzжсЃЈМДxyЦНУцЃЉЃЌжиЕўЧјЖдГЦЕиЗжУцдкЦНУцСНБпЃЌВЈКЏЪ§ЕФЗћКХЯрЗДЁЃетжжЖдГЦадГЦЮЊОЕУцЗДЖдГЦЃЌОпгаетжжЖдГЦадЕФЙВМлМќЮЊІаМќЁЃЭЌбљЃЌNдзгЕФ2py-2pyЙьЕРбиМќжсЗНЯђжиЕўЪБЃЌаЮГЩЕФвВЪЧІаМќЃЌЖдГЦУцЮЊxzЦНУцЁЃЫљвдN2ЗжзгжаЕФЙВМлВЮМќЃЌвЛИіЪЧІвМќЃЌСНИіЪЧІаМќЁЃ
,ЃЌЕБЫќУЧбиМќжсЗНЯђЯрЛЅППНќЪБЃЌ2px-2pxжиЕўГЪдВжљаЮЖдГЦЃЌаЮГЩвЛИіІвМќЃЛ2pz-2pzЙьЕРбиМќжсЗНЯђжиЕўЪБЃЌдђгавЛИіЖдГЦУцЁЃетИіЖдГЦУцЭЈЙ§МќжсЖјДЙжБгкzжсЃЈМДxyЦНУцЃЉЃЌжиЕўЧјЖдГЦЕиЗжУцдкЦНУцСНБпЃЌВЈКЏЪ§ЕФЗћКХЯрЗДЁЃетжжЖдГЦадГЦЮЊОЕУцЗДЖдГЦЃЌОпгаетжжЖдГЦадЕФЙВМлМќЮЊІаМќЁЃЭЌбљЃЌNдзгЕФ2py-2pyЙьЕРбиМќжсЗНЯђжиЕўЪБЃЌаЮГЩЕФвВЪЧІаМќЃЌЖдГЦУцЮЊxzЦНУцЁЃЫљвдN2ЗжзгжаЕФЙВМлВЮМќЃЌвЛИіЪЧІвМќЃЌСНИіЪЧІаМќЁЃ
ХфЮЛЙВМлМќЧАУцНщЩмЕФЙВМлМќЕФЙВгУЕчзгЖдЖМЪЧгЩВЮгыГЩМќЕФИїдзгЗжБ№ЬсЙЉЕФЪєгкЦеЭЈЙВМлМќЁЃЕЋЪЧгааЉЙВМлЗжзгжаЕФЙВМлМќВЂЗЧШчДЫЃЌШчЙћІвМќЕФЙВгУЕчзгЖдЪЧгЩЕЅЗНУцЬсЙЉЕФЃЌдђетРрЙВМлМќЖЈвхЮЊІвХфЮЛЙВМлМќЃЌМђГЦЮЊХфМлМќЁЃШчЙћІаМќЕФЙВгУЕчзгЖдгЩЕЅЮЛЗНУцЬсЙЉЃЌдђГЦЮЊІаХфМќЁЃР§ШчЃЌH+КЭNH3ЗДгІЩњГЩNH4+ЪБЃЌNH3ЗжзгжаNдзгЩЯЕФЙТЕчзгНЋЭЌЪБНјШыH+РызгжаФмСПзюЕЭЕФ1sПеЙьЕРЃЌаЮГЩвЛИіІвМќЃЌгЩгкетИіІвМќЕФГЩМќЕчзгЪЧгЩNдзгЕЅЗНУцЬсЙЉЕФЃЌЫљвдГЦЮЊІвХфЃЈЮЛЙВМлЃЉМќЃЌЦфЕчзгдЦЖдМќжсЪЧдВжљаЮЖдГЦЕФЁЃ
гжШчЃЌCOЗжзгЕФГЩМќЧщПіЃЌCдзгНщЕчзгВуЕФЛљЕчзгзщЬЌЮЊ![]() ,OдзгМлЕчзгВуЕФЛљЕчзгзщЬЌЮЊ2s22p4Лђ
,OдзгМлЕчзгВуЕФЛљЕчзгзщЬЌЮЊ2s22p4Лђ![]() ЃЌЕБЖўепЛЏКЯЪБЃКЖўепЕФ2px-2pxЕчзгдЦжиЕўГЩІвМќЃЈМќжсЮЊxжсЃЉЃЛ2py-2pyЕчзгдЦжиЕўГЩІаyМќЃЛOдзгЕФ
ЃЌЕБЖўепЛЏКЯЪБЃКЖўепЕФ2px-2pxЕчзгдЦжиЕўГЩІвМќЃЈМќжсЮЊxжсЃЉЃЛ2py-2pyЕчзгдЦжиЕўГЩІаyМќЃЛOдзгЕФ![]() ЕчзгНјШыCдзгЕФ
ЕчзгНјШыCдзгЕФ![]() ПеЙьЕРаЮГЩІаzХфМќЁЃ
ПеЙьЕРаЮГЩІаzХфМќЁЃ
дкЗжзгНсЙЙжаЃЌХфМќЭЈГЃвдЁАЁћЁБЗћКХБэЪОЃЌМ§ЭЗЦ№ЕуЮЊЬсЙЉЙТЖдЕчзгЕФдзгЃЌжеЕуЮЊЬсЙЉПеЙьЕРНгЪмЙТЖдЕчзгЕФдзгЁЃ
ЭЈЙ§вдЩЯЕФСНЬжТлПЩжЊЃЌаЮГЩХфМќБиаыЭЌЪБОпБИСНЩЯЬѕМўЃКЕквЛЃЌГЩМќдзгжагІгавЛИідзгжСЩйгавЛИіЙТЕчзгЖдЃЛЕкЖўЃЌГЩМќЕФСэвЛИідзгжаЃЌдкЦфМлЕчзгВужажСЩйгавЛИіПеЕФдзгЙьЕРЁЃХфМќЦеБщДцдкгкХфЮЛЛЏКЯЮяжаЃЌгаЙиХфМќЕФЮЪЬтНЋдкХфЮЛЛЏКЯЮяжаНјвЛВНЬжТлЁЃ
дзгЙьЕРЕФдгЛЏ(hybridization of atom orbitals)
ЙВМлМќРэТлШЗЪЕФмНтЪЭЙВМлЗжзгЕФГЩМќЙ§ГЬЃЌЕЋдкНтЪЭЗжзгМИКЮЙЙаЭЪБШДгіЕНСЫКмДѓРЇФбЃЌР§ШчCH4ЗжзгЕФМИКЮЙЙаЭ(geometric configuration)ЃЌИљОнЪЕбщВтЖЈЃЌ4ИіCЁЊHМќЕФМќГЄОљЯрЕШЃЌЮЊ109pmЃЌМќФмОљЮЊ414kJ/molЃЌCЁЊHМфЕФМаНЧОљЮЊ109Ёу28Ёф,ЫќУЧЙЙГЩвдCдзгЮЊжааФЕФе§ЫФУцЬхМИКЮЙЙаЭЃЌЖјИљОнМлМќРэТлШДЮоЗЈНтЪЭетИіЪТЪЕЃЌЖдH2OЗжзгКЭNH3ЗжзгЕФМИКЮЙЙаЭЕФНтЪЭЃЌвВгіЕНЯрЭЌЕФРЇФбЁЃ
ЕчзгХфЖдЗЈЕФЪЇАмЃЌЦфжївЊдвђЪЧЃЌВЛФмМђЕЅЕигУздгЩдзгЙьЕРДњЬцГЩМќКѓЗжзгжаЕФдзгЙьЕРЁЃЪЕМЪЩЯЃЌЕБдзгВЮгыЛЏбЇЗДгІЃЌВЂНсКЯГЩЗжзгЪБЃЌвбВЛдйЪЧздгЩдзгСЫЁЃдкетжжЧщПіЯТЃЌдзгжаЕФМлЕчзгВЛНівЊЪмздЩэжааФСІГЁЕФзїгУЃЌЛЙвЊЪмЕНгыЦфМќКЯдзгЕчГЁЕФзїгУЁЃвђДЫЃЌЕБгаМќКЯдзгЕчГЁзїгУЪБЃЌздгЩдзгМлЕчзгВужаЕФФГИіЕчзгЫљДІЕФзДЬЌЃЈдзгЙьЕРЃЉЃЌНЋЛсЗЂЩњИФБфЁЃЮЊДЫЃЌдкСПзгЛЏбЇжаНјааШчЯТДІРэЃКНЋздгЩдзгжаЕФдзгЙьЕРНјааЯпадзщКЯЃЈЕўМгЃЉВњЩњвЛИіаТЕФВЈКЏЪ§ЃЌгУЫќБэЪОГЩМќКѓЕФдзгЙьЕРЁЃетжжаТЕФВЈКЏЪ§ГЦЮЊдгЛЏдзгЙьЕРЃЌМђГЦдгЛЏЙьЕР(hybrid orbital)ЁЃетжжЯпадзщКЯЙ§ГЬГЦЮЊдзгЙьЕРдгЛЏ(atom orbital hybridization)ЁЃдкЗжзгAXmжаЃЌAдзгГЦЮЊжааФдзгЃЌXдзгГЦЮЊЖЫдзгЃЌЙьЕРдгЛЏвЛАуЗЂЩњдкжааФдзгжаЃЌЖјЖЫдзгВЛдгЛЏЁЃ
дгЛЏЙьЕРгаШчЯТЬиЕуЃК
ЃЈ1ЃЉФмСПЯрНќддђЃЌдквЛИідзгжаЃЌвЛАуЪЧжааФдзгМлЕчзгВуЕФФмСПЯрНќЕФдзгЙьЕРЃЌдкСэвЛИідзгЃЈЖЫдзгЃЉЕФзїгУЯТЃЌВХФмЗЂЩњдгЛЏЁЃжааФдзгжаФмСПЯрНќЃЌВЂФмВЮгыдгЛЏЕФдзгЙьЕРЃЌГЃМћЕФгавдЯТМИзщЃК
aзщЃКnsnpЃЌГЦЮЊs-pаЭдгЛЏЃЌКЌspЃЌsp2КЭsp3дгЛЏЃЛ
bзщЃК(nЃ1)dnsnpЃЌГЦd-s-pаЭдгЛЏЃЌКЌds2ЃЌd2sp3ЕШдгЛЏЃЛ
cзщЃКnsnpndГЦs-p-dаЭдгЛЏЃЌКЌsp3d2ЕШдгЛЏЁЃ
БОНкжиЕуНщЩмs-pаЭдгЛЏЁЃ
ЃЈ2ЃЉЙьЕРЪ§ФПЪиКуддђЃЌМДдзгжаВЮгыдгЛЏЕФдзгЙьЕРЪ§NЕШгкаЮГЩЕФдгЛЏЕРЪ§ЃЌР§ШчЃЌдкCH4ЗжзгжаЃЌCдзгга4ИідзгЙьЕРЃЈМД1Иі2sЁЂ3Иі2pЙьЕРЃЉаЮГЩЕФsp3дгЛЏЙьЕРЪ§вВЪЧ4ИіЁЃ
ЃЈ3ЃЉФмСПжиаТЗжХфддђЃЌдгЛЏЧАКѓЕФИїдзгЙьЕРОпгаШЗЖЈЕФФмСПЃЌЖјдгЛЏЃЈдзгЃЉЙьЕРЕФФмСПЃЌжЛгаЦНОљжЕЃЌВЂЧвИїдгЛЏЙьЕРФмСПЦНОљжЕПЩвдЯрЕШЃЈЕШаддгЛЏЃЉЃЌвВПЩВЛЯрЕШЃЈВЛЕШаддгЛЏЃЉЁЃетОіЖЈгкОпЬхЗжзгЕФЬѕМўЁЃ
ЃЈ4ЃЉзюДѓжиЕўддђЃЌдзгЙьЕРЕФдгЛЏПЩвдЬсИпЫќЕФГЩМќФмСІЃЌаЮГЩИќМгЮШЖЈЕФЙВМлЗжзгЁЃетЪЧвђЮЊдкКЫМфОрВЛБфЕФЧщПіЯТЃЌОЙ§дгЛЏЕФжааФдзгЕФЕчзгдЦгыМќКЯдзгЕчзгдЦЕФжиЕўЧјБШЮДдгЛЏЕФвЊДѓЕУЖрЃЌТњзуЕчзгдЦзюДѓжиЕўдРэЕФвЊЧѓЃЌвђЖјЬсИпСЫГЩМќФмСІЃЈМћЭМ9Ѓ5ЃЉЁЃ
ЃЈ5ЃЉЃЎдгЛЏЙьЕРЖдГЦадЗжВМддђЃЌИїИідгЛЏЙьЕРдкКЫЭтПеМфвЊВЩШЁзюЖдГЦЕФПеМфЗжВМЗНЪНЃЌвдЪЙГЩМќЕчзгжЎМфЕФХХГтСІзюаЁЁЃ
дгЛЏЙьЕРРраЭгыЗжзгПеМфМИКЮЙЙаЭ
ЃЈ1ЃЉspЕШаддгЛЏМАЗжзгЕФМИКЮЙЙаЭ
BeдзгЕФЛљЕчзгзщЬЌЮЊ1s22s2ЃЌдкBeCl2ЗжзгжаBeдзгЕФвЛИіnsЙьЕРКЭnpЙьЕРЃЈШ§ИіpЙьЕРжаЕФШЮвтвЛИіЃЉзщКЯЃЌЕУЕНСНИіЕШадspдгЛЏЙьЕРЃЌУПИідгЛЏЙьЕРжаОљКЌга1/2sГЩЗжКЭ1/2pГЩЗжЃЌСНИідгЛЏЙьЕРЕФМаНЧЮЊ180ЁуЃЌBeCl2ЗжзгГЪжБЯпаЭЁЃ(ЭМ9Ѓ5)
ЃЈ2ЃЉsp2ЕШаддгЛЏМАЗжзгЕФМИКЮЙЙаЭ
BдзгЕФЛљЕчзгзщЬЌ1s22s22p1ЃЌдкBF3ЗжзгжаBдзгЕФвЛИіnsЙьЕРКЭСНИіnpЙьЕРЃЈШ§ИіpЙьЕРжаЕФШЮвтСНИіЃЉзщКЯЃЌЕУЕНШ§ИіЕШадsp2дгЛЏЙьЕРЁЃЫљвдИїдгЛЏЙьЕРжа1/3sГЩЗжКЭ2/3pГЩЗжЃЌдгЛЏЙьЕРМфМаНЧЮЊ120ЁуЙЙГЩЦНУцШ§НЧаЮЗжзгЃЈМћЭМ9Ѓ6ЃЉЁЃ
ЃЈ3ЃЉsp3ЕШаддгЛЏМАЗжзгМИКЮЙЙаЭ
CдзгЕФЛљЕчзгзщЬЌЮЊ1s22s22p2ЁЃдкCH4ЗжзгжаЃЌCдзгЩЯЕФЕчзгЯШМЄЗЂЮЊ1s22s12p3ЃЌШЛКѓгЩвЛИіnsЙьЕРКЭШ§ИіnpЙьЕРзщЖјГЩsp3дгЛЏЙьЕРЁЃУПИідгЛЏЙьЕРКЌга1/4sГЩЗжКЭ3/4pГЩЗжЃЌГЪе§ЫФУцЬхЙЙаЭЁЃЃЈМћЭМ9Ѓ7ЃЉЁЃ
ЃЈ4ЃЉS-PаЭВЛЕШаддгЛЏМАЗжзгЙЙаЭ(molecularconfiguration)
ЕШаддгЛЏЛЙВЛФмНтЪЭЮЊЪ§жкЖрЕФЗжзгМИКЮЙЙаЭЖдЪЕбщНсЙЙЕФЦЋРыЁЃЯТУцвдNH3ЗжзгЮЊР§РДЫЕУїетжжЦЋРыВњЩњЕФдвђЁЃ
NМлЕчзгВуЕФЛљЕчзгзщЬЌЮЊ2s22p3ЃЌМД![]() ЁЃNH3ЗжзгЕФжааФдзгNВЩШЁsp3дгЛЏЃЌгЩдгЛЏЧАКѓЕФФмМЖЗжВМЭМЃЈШчЭМ9Ѓ8ЃЉПЩвдЗЂЯжЃКЫФИіsp3дгЛЏЙьЕРЕФФмСПВЂВЛЯрЭЌЃЌЦфжаБЛВЛВЮгыГЩМќЕФЙТЖдЕчзгеМОнЕФвЛИідгЛЏЙьЕРЕФФмСПБШЦфЫћШ§ИіЕЭЁЃСПзгЛЏбЇМЦЫуКЭКЫДХЙВеёЪЕбщНсЙћБэУїЃЌЧАепЕФsГЩЗжЮЊ79%ЃЌpГЩЗжЮЊ21%ЃЌКѓепЕФsГЩЗжЮЊ7%ЃЌpГЩЗжЮЊ93%ЁЃгкЪЧЮвУЧНЋИїдгЛЏЙьЕРжаЕФЭЌУћдзгЙьЕРЕФГЩЗжВЛЭъШЋЕШЭЌЕФдгЛЏГЦВЛЕШаддгЛЏЁЃ
ЁЃNH3ЗжзгЕФжааФдзгNВЩШЁsp3дгЛЏЃЌгЩдгЛЏЧАКѓЕФФмМЖЗжВМЭМЃЈШчЭМ9Ѓ8ЃЉПЩвдЗЂЯжЃКЫФИіsp3дгЛЏЙьЕРЕФФмСПВЂВЛЯрЭЌЃЌЦфжаБЛВЛВЮгыГЩМќЕФЙТЖдЕчзгеМОнЕФвЛИідгЛЏЙьЕРЕФФмСПБШЦфЫћШ§ИіЕЭЁЃСПзгЛЏбЇМЦЫуКЭКЫДХЙВеёЪЕбщНсЙћБэУїЃЌЧАепЕФsГЩЗжЮЊ79%ЃЌpГЩЗжЮЊ21%ЃЌКѓепЕФsГЩЗжЮЊ7%ЃЌpГЩЗжЮЊ93%ЁЃгкЪЧЮвУЧНЋИїдгЛЏЙьЕРжаЕФЭЌУћдзгЙьЕРЕФГЩЗжВЛЭъШЋЕШЭЌЕФдгЛЏГЦВЛЕШаддгЛЏЁЃ
вЛАуЧщПіЯТЃЌЗВЪЧжааФдзгдкГЩМќжЎКѓЃЌЦфМлЙьЕРЩЯШдгаЙТЖдЕчзгЪБЃЌЭЈГЃШЁВЛЕШаддгЛЏЁЃГЩМќдгЛЏЙьЕРжаЃЌs(p)ГЩЗждНДѓЃЈаЁЃЉЃЌМќНЧдНДѓЃЛетОЭНтЪЭСЫNдзгЫфШЛШЁsp3дгЛЏЃЌЖјМќНЧаЁгк109Ёу28ЁфЃЌЮЊ107Ёу20ЁфЕФдвђЁЃЭЌРэПЩвдЖЈадЫЕУїЃЌH2OЗжзгЕФМќНЧЮЊЪВУДМШВЛЪЧ90ЁуЃЌвВВЛЪЧ109Ёу28ЁфЃЌЖјЪЧ104Ёу45ЁфЕФдвђЁЃгІЕБзЂвтЖдгкЭЌжждзгдкВЛЭЌЗжзгжаЃЌЛђдкЭЌвЛЗжзгжаЕФВЛЭЌЮЛжУЃЌЫќУЧЫљШЁЕФдгЛЏаЮЪНВЛЭъШЋЯрЭЌЁЃР§ШчЃЌдкNH3ЁЂNH4+ЁЂNO2ЁЂNO2+КЭNOЗжзгЃЌРызгЯЕСажаЃЌNдзгЫљВЩШЁЕФдгЛЏаЮЪНОЭИїВЛЯрЭЌЃЛгжР§ШчЃЌ![]() дкЗжзгжаЕФСНИіCдзгКЭСНИіOдзг,ЫљВЩШЁЕФдгЛЏаЮЪНвВЪЧИїВЛЯрЭЌЁЃЫљвдвЛИідзгдкЗжзгжаОПОЙгІИУШЁЪВУДбљЕФдгЛЏаЮЪНЃЌгІИљОнОпЬхЧщПіРДШЗЖЈЁЃ
дкЗжзгжаЕФСНИіCдзгКЭСНИіOдзг,ЫљВЩШЁЕФдгЛЏаЮЪНвВЪЧИїВЛЯрЭЌЁЃЫљвдвЛИідзгдкЗжзгжаОПОЙгІИУШЁЪВУДбљЕФдгЛЏаЮЪНЃЌгІИљОнОпЬхЧщПіРДШЗЖЈЁЃ
МлВуЕчзгЖдЛЅГтРэТлЃЈvalenceshellpairrepulsiontheoryVSEPRTЃЉдкдЄВтвЛаЉЗжзгЛђРызгЕФПеМфЙЙаЭЗНУцБШНЯМђБуЃЌЫќгыдгЛЏЙьЕРРэТлХфКЯЃЌФмЗНБуЕиНтЪЭвЛАуЗжзгЛђРызгЕФГЩМќЧщПіКЭПеМфЙЙаЭЁЃХаЖЯЗжзгЛђРызгЕФПеМфЙЙаЭОЭЪЧвдетаЉЕчзгЖдЕФПеМфХХВМЮЊвРОнЁЃвЛИіЖрдзгЙВМлаЭЗжзгAbnЛђРызг![]() ЕФПеМфЙЙаЭЃЌШЁОігкжааФдзгAжмЮЇЕФГЩМќЕчзгЖд(bonded electron pair)КЭЙТЕчзгЖд(lone electron pair)ЕФЪ§ФПЃЛЕчзгЖджЎМфЕФЯрЛЅХХГтЃЌЪЙетаЉЕчзгЖдЛЅЯрДІгкОЁПЩФмдЖРыЕФЮЛжУЩЯвдЪЙГтСІзюаЁЃЛЗжзгПеМфЙЙаЭЕФвЛАуХаЖЯЗНЗЈЪЧЃК
ЕФПеМфЙЙаЭЃЌШЁОігкжааФдзгAжмЮЇЕФГЩМќЕчзгЖд(bonded electron pair)КЭЙТЕчзгЖд(lone electron pair)ЕФЪ§ФПЃЛЕчзгЖджЎМфЕФЯрЛЅХХГтЃЌЪЙетаЉЕчзгЖдЛЅЯрДІгкОЁПЩФмдЖРыЕФЮЛжУЩЯвдЪЙГтСІзюаЁЃЛЗжзгПеМфЙЙаЭЕФвЛАуХаЖЯЗНЗЈЪЧЃК
1ЃЎШЗЖЈдкжааФдзгAМлЕчзгВужаЕФЕчзгЖдзмЪ§
АбжааФдзгAдгаЕФМлЕчзгЪ§МгЩЯХфЮЛдзгBЬсЙЉЕФЕчзгЪ§МДЕУМлЕчзгВужаЕФзмЕчзгЪ§ЁЃдке§ГЃЕФЙВМлМќжаЃЌЙцЖЈЃК
ЃЈ1ЃЉВЛЭЌдЊЫиЕФдзгДІгкВЛЭЌЕиЮЛЪБЃЌЬсЙЉСЫМлВуЕчзгЪ§ВЛЭЌЁЃЧтКЭТБЫидзгИїЬсЙЉ1ИіМлЕчзгЃЛЕЋЕБТБЫидзгзїЮЊжааФдзгЪБЃЌдђЬсЙЉ7ИіМлЕчзгЁЃбѕзхдЊЫизїЮЊЖЫдзгЪБПЩШЯЮЊВЛЬсЙЉЙВгУЕчзгЃЛЕЋЕБбѕзхдЊЫиЕФдзгзїЮЊжааФдзгЪБЃЌдђЬсЙЉ6ИіМлЕчзгЁЃетЪЧвђЮЊЃКдзгзїЮЊХфЮЛдзгдкГЩМќЪБЃЌПеГі1Иі2pЙьЕРНгЪмжааФдзгЬсЙЉЕФЙТЕчзгЖдЖјаЮГЩХфЮЛМќЃЌ
ЃЈ2ЃЉШчЙћЫљЬжТлЕФЪЧЖрдзгРызгЃЌдђвЊМгЩЯЛђМѕШЅгыЕчКЩЯрЖдгІЕФЕчзгЪ§ЁЃР§ШчЃЌ![]() РызгжаЕФPгІМгЩЯ3ИіЕчзгЃЌЖј
РызгжаЕФPгІМгЩЯ3ИіЕчзгЃЌЖј![]() РызгжаЕФNгІМѕШЅ1ИіЕчзгЁЃ
РызгжаЕФNгІМѕШЅ1ИіЕчзгЁЃ
ЃЈ3ЃЉШчЙћЫљЬжТлЕФЗжзгжаКЌгаЫЋМќЛђШ§МќЃЌдђАбЫќУЧЕБзі1ИіЕЅМќМДЮЊзїЮЊ2ИіЕчзгПДД§ЁЃвђЮЊдкЖржиМќЖдЗжзгаЮзДВЛЦ№ОіЖЈзїгУЁЃ
ЃЈ4ЃЉАбжааФдзгAЕФМлЕчзгВужаЕФзмЕчзгЪ§Г§вд2дђЕУЕНЕчзгЖдЪ§ЃЌШчгігаВЛГЩЖдЕФЕчзгЃЌвВзїЮЊЕчзгЖдПДД§ЁЃ
2ЃЎШЗЖЈЕчзгЖдЕФМИКЮЙЙаЭ
гЩжааФдзгAЕФМлЕчзгЪ§ЖдЃЌИљОнХХГтСІзюаЁЕФддђЃЌПЩвдШЗЖЈЕчзгЖдЕФРэЯыМИКЮЙЙаЭЁЃЖдгІгкЕчзгЖдЪ§ЮЊ2ЁЂ3ЁЂ4ЁЂ5ЁЂ6ЪБЃЌМИКЮЙЙаЭЗжБ№ЮЊжБЯпаЭЁЂЦНУцШ§НЧаЮЁЂе§ЫФУцЬхаЭЁЂШ§НЧЫЋзЖаЭКЭе§АЫУцЬхаЭЁЃ
3ЃЎШЗЖЈЗжзгЕФЮШЖЈНсЙЙ
ИљОнЕчзгЖдЕФМИКЮЙЙаЭЃЌАбХфЮЛдзгХХдкжааФдзгAЕФжмЮЇЃЌУПвЛЖдЕчзгСЌНг1ИіХфЮЛдзгЃЌЪЃЯТЕФЮДНсКЯЕФЕчзгЖдБуЪЧЙТЕчзгЖдЁЃПМТЧвдЯТвђЫиЃЌШЗЖЈЗжзгЙЙаЭЃЌЪЙЗжзгЕФФмСПзюЕЭЃК
ЃЈ1ЃЉПМТЧЙТЕчзгЖдЁЂГЩМќЕчзгЖдЖдЗжзгМИКЮЙЙаЭЕФгАЯь
ИљОнЙТЕчзгЖдЁЂГЩМќЕчзгЖдетМфЕФЯрЛЅГтСІЕФДѓаЁЫГађЃКЙТЕчзгЖд-ЙТЕчзгЖдЃОЙТЕчзгЖд-ГЩМќЕчзгЖдЃОГЩМќЕчзгЖд-ГЩМќЕчзгЖдЃЌШЗЖЈГтСІзюаЁЕФХХВМЮЊЮШЖЈНсЙЙЃЌВЂЙРМЦетжжНсЙЙЖдРэЯыМИКЮЙЙаЭЕФЦЋРыГЬЖШЁЃ
ЃЈ2ЃЉПМТЧжиМќЖдЗжзгМИКЮЙЙаЭЕФгАЯь
ШчЙћЗжзгжагажиМќЃЌЫфШЛжиМќжЛЫувЛИіЕчзгЖдЃЌЕЋЫќБЯОЙгаНЯЖрЕФМлЕчзгЃЌдкжааФдзгжмЮЇеМгаНЯДѓЕФПеМфЃЌвђЖјЖдИННќЕФЦфЫћЕчзгЖдгаНЯДѓЕФХХГтзїгУЁЃжиМќХХГтзїгУЕФДѓаЁЫГађЮЊЃКШ§жиМќЃОЫЋМќЃОЕЅМќЃОЕЅЕчзгЁЃвђДЫЃЌЫфШЛжиМќЛђІаМќВЛЛсИФБфЗжзгЕФЛљБОаЮзДЃЌЕЋЖдМќНЧвВЛсгавЛЖЈЕФгАЯьЁЃвЛАуРДЫЕЃЌКЌЕЅМќЕФМќНЧаЁЃЌКЌЫЋМќЕФМќНЧТдДѓЁЃР§ШчC2H4ЗжзгКЭCH2OЗжзгжаЃЌМќНЧЁЯHCCКЭЁЯHCOТдДѓгк120ЁуЖјМќНЧЁЯHCHТдаЁгк120ЁуЃЈМћЭМ9-10ЃЉЁЃ
ЃЈ3ЃЉПМТЧЕчИКадЖдЗжзгМИКЮЙЙаЭЕФгАЯь
жааФдзгКЭМќКЯдзгЕФЕчИКадЯрЖдДѓаЁЃЌвВЛсгАЯьЗжзгМИКЮЙЙаЭЃЈМќНЧДѓаЁЃЉЁЃЖдгкЭЌвЛжааФдзгЃЌгыЦфМќКЯЕФдзгЕФЕчИКаддНДѓЃЌЮќв§МќЖдЕчзгЕФФмСІОЭдНДѓЃЌМќЖдЕчзгдЦНЋМќКЯдзгЗНЯђвЦЖЏЁЃЖдИННќЦфЫћЕчЖдЕФХХГтзїгУОЭБШНЯаЁЃЌвђЖјМќНЧБфаЁЁЃР§ШчдкNH3КЭNF3ЗжзгжаЃЌx(F)ЃОx(N)ЃЌЕчИКадЃЌЫљвдЖўепЕФМќНЧЮЊЁЯFNFЃЈ102.1ЁуЃЉЃМЁЯHNH(107Ёу20Ёф)ЁЃЕБМќКЯдзгЯрЭЌЖјжааФдзгВЛЭЌЪБЃЌдђжааФдзгЕчИКаддНДѓЃЌМќЖдЕчзгдЦНЋЯђжааФдзгвЦЖЏЃЌВњЩњНЯДѓЕФХХГтзїгУЃЌМќНЧНЋБфДѓЁЃР§ШчдкH2XЃЈX=OЃЌSЃЌSeЃЌTeЃЉЗжзгжаЃЌжааФдзгЕФИКЕчадxЗжБ№ЮЊ3.44ЁЂ2.58ЁЂ2.55КЭ2.1ЃЌвђДЫЫќУЧЯргІЕФМќНЧЁЯHXHЗжБ№ЮЊ104.7ЁуЁЂ92.2ЁуЁЂ91.0ЁуЁЂ89.5ЁуЁЃ
ЃЈ4ЃЉМлВуЕчзгЖдЛЅГтРэТлХаЖЯЗжзгМИКЮЙЙаЭЕФЪЕР§
Р§[2Ѓ1]ХаЖЯNO2ЗжзгЕФМИКЮЙЙаЭЁЃ
НтЃКNдзгга5ИіМлЕчзгЃЌИљОнЩЯЪіЙцдђЃЌбѕдзгВЛЬсЙЉМлЕчзгЃЌвђДЫжааФдзгNЕФМлВуЕчзгзмЪ§ЮЊ5ЃЌЯрЕБгк3ИіЕчзгЖдЁЃNдзгМлВуЕчзгЖдЕФХХВМгІЮЊЦНУцШ§НЧаЭЃЌЫљвдNO2ЗжзгЕФНсЙЙЮЊЁАVЁБзжаЮЃЌЁЯONOРэТлжЕЮЊ120ЁуЃЌЕЋдкЙТЕчзгЖдЕФЮЛжУЪЕМЪЩЯжЛга1ИіЕчзгЃЌХХГтСІМѕаЁЪЙМќНЧвЊЩдДѓвЛаЉЪЕМЪЩЯЁЯONO=135ЁуЃЌШчЭМ9Ѓ10ЫљЪОЁЃ
МлМќРэТлЃЈАќКЌдгЛЏЙьЕРРэТлЃЉФмЫЕУїаэЖрЗжзгЕФаЮГЩЙ§ГЬКЭМИКЮЙЙаЭЃЌПЩЪЧгІгУЫќРДДІРэФГаЉЗжзгШчO2ЗжзгЕФНсЙЙЪБШДгіЕНСЫУЌЖмЁЃР§ШчЃЌАДМлМќРэТлЃЌO2ЗжзгЕФНсЙЙгІЮЊЁУ![]() ЁУЃЌЗжзгФкВПЕФЕчзгЖМвбХфЖдЃЌЕЋИљОнДХадЪЕбщЃЌВтЕУбѕЗжзгЮЊЫГДХадЮяжЪЃЌЫЕУїбѕЗжзгжаДцдкЮДГЩЖдЕчзгЁЃеташвЊгУЗжзгЙьЕРРэТлМгвдЫЕУїЁЃ
ЁУЃЌЗжзгФкВПЕФЕчзгЖМвбХфЖдЃЌЕЋИљОнДХадЪЕбщЃЌВтЕУбѕЗжзгЮЊЫГДХадЮяжЪЃЌЫЕУїбѕЗжзгжаДцдкЮДГЩЖдЕчзгЁЃеташвЊгУЗжзгЙьЕРРэТлМгвдЫЕУїЁЃ
ЗжзгЙьЕРРэТл(moleculeorbitaltheory)ЧПЕїЗжзгЕФећЬхадЃЌШЯЮЊдзгаЮГЩЗжзгвдКѓЃЌЕчзгВЛдйОжЯогкУПИідзгЕФдзгЙьЕРЃЌЖјЪЧдкећИіЗжзгЗЖЮЇФкдЫЖЏЁЃЗжзгжаУПИіЕчзгЕФдЫЖЏзДЬЌЃЌЭЌбљПЩгУЯргІЕФВЈКЏЪ§(wavefunction)ІзРДУшЪіЃЌетжжУшЪіЗжзгжаЕчзгдЫЖЏзДЬЌЕФВЈКЏЪ§НазіЗжзгЙьЕРЁЃвдЯТгУЭЌКЫЫЋдзгЗжзгЮЊР§ЫЕУїЗжзгЙьЕРРэТлЕФвЊЕуЁЃЗжзгЙьЕРЪЧгЩЗжзгжаИїИідзгЕФдзгЙьЕРЪЪЕБзщКЯаЮГЩЕФЁЃЗжзгЙьЕРЕФЪ§ФПЕШгкВЮМгзщКЯЕФШЋВПдзгЙьЕРЪ§ЁЃСПзгСІбЇ(quantummechanics)жЄУїЃЌгЩдзгЙьЕРзщКЯГЩЗжзгЙьЕРЪБЃЌаыТњзуЯТСаШ§ИіЬѕМўЃК
ЕквЛЃЌФмСПЯрНќЬѕМўЃКжЛгаФмСПЯрНќдзгЙьЕРВХФмгааЇЕизщКЯГЩЗжзгЙьЕРЁЃЮЊДЫЃЌдкЗжзгЙьЕРЗћКХжааыгУЯТБъзЂУїЖдгІЕФдзгЙьЕРЕФУћГЦЁЃ
ЕкЖўЃЌдзгЙьЕРЕФзюДѓжиЕўЬѕМўЃКвЊЧѓзщГЩЗжзгЙьЕРЕФИїИідзгЙьЕРОЁПЩФмЖрЕижиЕўЁЃИљОндзгЙьЕРЯрЛЅжиЕўЕФГЬЖШДѓаЁЃЌПЩвдНЋЗжзгЙьЕРЗжЮЊІвЗжзгЙьЕРКЭІаЗжзгЙьЕРЁЃР§ШчЃЌгЩsКЭsЙьЕРЁЂPxКЭPxЙьЕРзщКЯЫљаЮГЩЕФЗжзгЙьЕРЃЌЦфаЮзДЪЧбизХСНКЫСЌЯпЗНЯђГЪдВжљаЮЖдЯњЃЌетжжЗжзгЙьЕРНазіІвЗжзгЙьЕРЃЛгЩPyКЭPyЛђPzКЭPzЙьЕРзщКЯЫљаЮГЩЕФЗжзгЙьЕРЁЃгавЛИіЭЈЙ§КЫМфСЌЯпЕФЖдГЦНкУцЃЌГЪОЕУцЗДЖдГЦЃЌНазіІаЗжзгЙьЕРЃЈМћЭМ9Ѓ13ЃЉЁЃ
ЕкШ§ЃЌЖдГЦадЦЅХфЬѕМўЁЃ
дзгЙьЕРзщКЯГЩЗжзгЙьЕРЪБЃЌгаСНжжзщКЯЗНЪНЃКдзгЙьЕРЕФЭЌКХВПЗжжиКЯЛђвьКХЖМЗжжиКЯЖдГЦадВЛЦЅХфЃЌаЮГЩЗДМќЗжзгЙьЕРЁЃгыдРДЕФСНИідзгЙьЕРЕФФмМЖЯрБШНЯЃЌГЩМќЗжзгЙьЕР(bondingmolecularorbital)жаСНКЫМфЕчзгдЦУмЖШдіДѓЃЌЦфФмМЖНЕЕЭЃЛЗДМќЗжзгЙьЕР(antibondingmolecularorbital)жаСНКЫМфЕчзгдЦУмЖШМѕаЁЃЌФмМЖЩ§ИпЁЃГЩМќЙьЕРНЕЕЭЕФФмСПКЭЗДМќЗжзгЙьЕРЩ§ИпЕФФмСПЯрЕШЁЃЧАепГЦЮЊЖдГЦадЦЅХф(symmetricmatch)аЮГЩГЩМќЙьЕРЃЌКѓепГЦЮЊІвГЩМќЙьЕРКЭЕФЗДМќЙьЕРЗжБ№гУІвКЭІв*БэЪОЃЌІаГЩМќЙьЕРКЭЗДМќЙьЕРЃЌЗжБ№гУІаКЭІа*БэЪОЃЈЭМ9Ѓ13ЃЉЁЃгЩЭМПЩМћЃЌгЩs-sзщКЯжЛФмаЮГЩ1ИіІвsЙьЕРКЭ1Иі![]() ЙьЕРЃЌЖјpЃpзщКЯЃЌГ§аЮГЩ1Иі
ЙьЕРЃЌЖјpЃpзщКЯЃЌГ§аЮГЩ1Иі![]() ЙьЕРКЭ1Иі
ЙьЕРКЭ1Иі![]() ЙьЕРЭтЃЌЛЙПЩаЮГЩФмСПЯрЕШЃЌЛЅЯрДЙжБЕФ
ЙьЕРЭтЃЌЛЙПЩаЮГЩФмСПЯрЕШЃЌЛЅЯрДЙжБЕФ![]() КЭ
КЭ![]() ЗжзгЙьЕРвдМА
ЗжзгЙьЕРвдМА![]() КЭ
КЭ![]() ЗжзгЙьЕРЃЌЙВ6ИіЗжзгЙьЕРЁЃдкЭМ9-14жаНЋетаЉЗжзгЙьЕРАДФмСПИпЕЭХХСаГЩЗжзгЙьЕРФмМЖЭМЃЌЭМжаУПИљЖЬКсЯпДњБэ1ИіЗжзгЙьЕРЁЃ
ЗжзгЙьЕРЃЌЙВ6ИіЗжзгЙьЕРЁЃдкЭМ9-14жаНЋетаЉЗжзгЙьЕРАДФмСПИпЕЭХХСаГЩЗжзгЙьЕРФмМЖЭМЃЌЭМжаУПИљЖЬКсЯпДњБэ1ИіЗжзгЙьЕРЁЃ
дкЗжзгЙьЕРЩЯЬюГфЕчзгМДЙЙГЩЗжзгЙьЕРЕчзгЗжВМЪНЁЃЕчзгдкЗжзгЙьЕРжаЕФЗжВМКЭЕчзгдкдзгЙьЕРжаЕФЗжВМвЛбљЃЌЗўДгХнРћВЛЯрШндРэЁЂзюЕЭФмСПдРэКЭКщЬиЙцдђЁЃИљОнЗжзгЙтЦзЕФЪЕбщЪ§ОнШЗЖЈЕФЗжзгЙьЕРФмМЖИпЕЭЕФЫГађЃЌАДееЕчзгЗжВМЕФддђЃЌПЩвдаДГіЗжзгЕФЕчзгЗжВМЪНЁЃЯжОйР§ЫЕУїШчЯТЃК
ЕкЖўжмЦкдЊЫиЭЌКЫЫЋдзгЗжзгЕФЗжзгЙьЕРФмМЖЫГађгаСНжжРраЭЃЌO2ЁЂF2ЮЊвЛРрЃЌвдO2ЮЊДњБэЃЛLi2ЁЋN2ЮЊСэвЛРрЃЌвдN2ЮЊДњБэЁЃ
O2ЗжзгЙВга16ИіЕчзгЃЌЦфжа12ИіЮЊЭтВуЕчзгЁЃАДЕчзгЗжВМддђКЭЭМ9Ѓ17ЕФФмМЖЫГађЃЌЦфЕчзгЗжВМЪНПЩаДГЩЃК
![]() ЃЈ9-1ЃЉ
ЃЈ9-1ЃЉ
ЪНжаЯТЛЎЯпБэЪОФмСПМђВЂ(energydegeneration)ЕФЙьЕРЁЃ
N2ЗжзгЙВга14ИіЕчзгЃЌЦфжа10ИіЮЊЭтВуЕчзгЃЌАДЕчзгЗжВМддђЃЌЦфЕчзгЗжВМЪНПЩаДГЩЃК
![]() ЃЈ9-2ЃЉ
ЃЈ9-2ЃЉ
ЪНжаKKЕШаЇгк![]() ЃЌБэЪОСНИіЕЊдзгЕФKВуШЋГфТњЁЃ
ЃЌБэЪОСНИіЕЊдзгЕФKВуШЋГфТњЁЃ
БШНЯO2ЗжзггыN2ЗжзгЕФЗжзгЙьЕРФмМЖЫГађЗЂЯжЃЌЫќУЧЕФЧјБ№дкгкЃЌ![]() ФмМЖЕФЮЛжУЗЂЩњБфЛЏЁЃдкO2ЗжзгжаЃЌ
ФмМЖЕФЮЛжУЗЂЩњБфЛЏЁЃдкO2ЗжзгжаЃЌ![]() ЕФФмМЖБфЕУЕЭгк
ЕФФмМЖБфЕУЕЭгк![]() МА
МА![]() ЁЃЩЯЪіЗжзгЙьЕРЕчзгЗжВМЪНЃЌЪЧЗжзгДІгкЛљЬЌЪБЕФЕчзгНсЙЙЃЌЙЪгжГЦЮЊЗжзгЕФЛљЕчзгзщЬЌЁЃ
ЁЃЩЯЪіЗжзгЙьЕРЕчзгЗжВМЪНЃЌЪЧЗжзгДІгкЛљЬЌЪБЕФЕчзгНсЙЙЃЌЙЪгжГЦЮЊЗжзгЕФЛљЕчзгзщЬЌЁЃ
ИљОнЗжзгЕФЛљЕчзгзщЬЌЃЌПЩвдКмЗНБуЕиЬжТлЗжзгЕФГЩМќЧщПіЁЂЗжзгЕФДХадЃЌВЂЛГіЗжзгЕФНсЙЙЪНЁЃ
(1) МќМЖ(bondorder)
ЗжзгЙьЕРРэТлжаЃЌЙВМлМќЕФЧПЖШгУМќМЖБэЪОЁЃМќМЖАДЯТЪНМЦЫуЃК
![]() ЃЈ9-3ЃЉ
ЃЈ9-3ЃЉ
ЕБГЩМќЙьЕРКЭЗДМќЙьЕРЕФЕчзгЪ§ЯрЕШЪБЃЌМќаЇгІгыЗДМќаЇгІЛЅЯрЕжЯћЃЌЖдГЩМќУЛгаЙБЯзЃЌжЛгаОЛГЩМќЕчзгЪ§ЖдГЩМќВХгаЙБЯзЃЌМќМЖдНДѓЃЌМќЕФЧПЖШдНДѓЃЌЗжзгдНЮШЖЈЁЃ
ЃЈ2ЃЉЗжзгЕФДХад
ЮяжЪЕФДХадЪЧжИЮяжЪдкДХГЁжаБэЯжГіРДЕФаджЪЁЃвЛАуЮяжЪЕФДХаджївЊгЩЕчзгдЫЖЏРДБэЯжЃЌЫќКЭдзгЁЂЗжзгЛђРызгЕФЮДГЩЕчзгЪ§гажБНгЕФЙиЯЕЃЌШєЗжзгЛђРызгжаЫљгаЕФЕчзгЖМвбХфЖдЃЌдђетжжЮяжЪжУгкДХГЁжаЛсБЛгеЕМГівЛИігыЭтДХГЁЗДЯђЕФаЁДХОиЃЌДгЖјЯїШѕЭтДХГЁЕФЁЃетжжЮяжЪГЦЮЊЗДДХадЮяжЪЁЃШєЗжзгЛђРызгжаДцдкЮДГЩЖдЕчзгЃЌдкЭтДХГЁзїгУЯТЃЌЛсБЛДХГЁЮќв§ЃЌДгЖјМгЧПЭтДХГЁЕФЧПЖШЃЌетжжЮяжЪГЦЮЊЫГДХадЮяжЪЁЃЭЈГЃАбЫГДХадЮяжЪдкДХГЁжаВњЩњЕФДХаЇгІгУДХОиІЬmРДБэЪОЁЃІЬmЕФЕЅЮЛЮЊВЃЖћДХзгІЬBЁЃ
ЗжзгЕФЫГДХадМИКѕШЋВПгЩЕчзгЕФзда§ЫљЙБЯзЃЌЦфДХОиІЬmгыЮДГЩЖдЕчзгЪ§ЕФЙиЯЕЮЊЃК
![]() ЃЈ9-4ЃЉ
ЃЈ9-4ЃЉ
ЪНжаnЮЊЗжзгЮДГЩЖдЕчзгЪ§ЃЌІЬBЮЊДХОиЕЅЮЛЃЌГЦЮЊВЃЖћДХзгЃЈІЬB=9.27ЁС10Ѓ24AЁЄm2ЃЉЁЃO2Зжзгжага2ИіЮДГЩЖдЕчзгЃЈn=2ЃЉЃЌЦфДХОиЮЊЃК
![]()
N2ЗжзгжаУЛгаЮДГЩЖдЕчзгЃЈn=0ЃЉЃЌЦфДХОиЮЊ0ЃЌгыЪЕбщНсЙћЗћКЯЁЃ
МќФм(bondenergy)ЁЂМќГЄ(bondlength)КЭМќНЧ(bondangle)ЪЧБэЪОЙВМлМќаджЪЕФживЊЮяРэСПЃЌЭГГЦЮЊМќВЮЪ§(bondparameter)ЁЃМШШЛЗжзгЪЧгЩдзгЭЈЙ§ЙВМлМќНсКЯЖјГЩЃЌвђДЫЙВМлМќЕФаджЪОЭОіЖЈСЫЗжзгЕФаджЪЁЃР§ШчЃКЙВМлМќгаБЅКЭадЃЌОЭОіЖЈСЫЗжзгОпгаШЗЖЈЕФзщГЩЃЈгУЗжзгЪНБэЪОЃЉЃЛЙВМлМќЕФЗНЯђадЃЌОЭОіЖЈСЫЗжзгОпгаШЗЖЈЕФПеМфМИКЮЙЙаЭЃЈгУМќГЄКЭМќНЧБэЪОЃЉЃЛЙВМлМќгаЧПШѕВюБ№ЃЌОЭОіЖЈСЫЗжзгЕФЮШЖЈадЕФВювьЃЈгУМќФмБэЪОЃЉЁЃЫљвдМќВЮЪ§ЪЧУшЪіЗжзгаджЪЕФживЊВЮЪ§ЁЃ
МќФмЃКдкБъзМзДЬЌЯТЃЌЦјЬЌЮяжЪABЖЯПЊ1molМќаЮГЩжаадЦјЬЌЮяжЪAЃЌBЫљашвЊЕФФмСПГЦЮЊAЁЊBМќЕФМќФм(bondenergy)ЁЃетИіФмСПЕШгкИУЙ§ГЬЕФБъзМьЪБфЃЌМќФмЪЧКтСПЛЏбЇМќФмЕФЮяРэСПЃЌЗћКХЮЊEЃЌЕЅЮЛЮЊkJ/molЃЌМќФмЕФЖЈвхжївЊгУгкКтСПЙВМлМќЕФЧПЖШЁЃ
ЖдгкЫЋдзгЗжзгЃЌМќФмЕШгкЗжзгЕФРыНтФмDЁЃ
МќГЄЃКЗжзгФкГЩМќСНдзгКЫМфЕФЦНКтОрРыГЦЮЊМќГЄЃЈBondlengthЃЉЁЃМќГЄПЩвдгУЗжзгЙтЦзЛђXЩфЯпбмЩфЗНЗЈВтЕУЁЃ
ДгЗжЮіДѓСПЪЕбщЪ§ОнЗЂЯжЃЌЭЌвЛжжМќдкВЛЭЌЗжзгжаЕФМќГЄЪ§жЕЛљБОЩЯЪЧИіЖЈжЕЁЃетЫЕУївЛИіМќЕФаджЪжївЊШЁОігкГЩМќдзгЕФБОадЁЃСНИіШЗЖЈЕФдзгжЎМфЃЌШчЙћаЮГЩВЛЭЌЕФЛЏбЇМќЃЌЦфМќГЄГЩЖЬЃЌМќФмОЭдНДѓЃЌМќОЭдНРЮЙЬЁЃ
МќНЧЃКдкЗжзгжаСНИіЯрСкЛЏбЇМќжЎМфЕФМаНЧГЦЮЊМќНЧ(bondangle)ЁЃЯёМќГЄвЛбљЃЌМќНЧЪ§ОнПЩвдгУЗжзгЙтЦзЛђXЩфЯпбмЩфЗЈВтЕУЁЃШчЙћжЊЕРСЫФГЗжзгФкШЋВПЗжбЇМќЕФМќГЄКЭМќНЧЪ§ОнЃЌФЧУДетИіЗжзгЕФМИКЮЙЙаЭОЭШЗЖЈСЫЁЃПЩМћЃЌМќНЧКЭМќГЄЪЧУшЪіЗжзгМИКЮНсЙЙЕФСНИівЊЧѓЁЃ
ЕкАЫеТ ХфКЯЮяЕФНсЙЙКЭаджЪ
НёЬьЃЌЮвУЧвЛЦ№РДбЇЯАХфКЯЮяЕФНсЙЙгыаджЪЁЃХфКЯЮяЪЧLewisЫсМюЕФМгКЯЮяЁЃР§ШчЃК[Ag(NH3)2]+ЪЧLewisЫсAg+КЭNH3ЕФМгКЯЮяЁЃLewisЫсГЦЮЊаЮГЩЬх(жааФРызгЃЉЃЛLewisМюГЦЮЊХфЮЛЬхЁЃаЮГЩЬхгывЛЖЈЪ§ФПЕФХфЮЛЬхвдХфЮЛМќАДвЛЖЈЕФПеМфЙЙаЭНсКЯЕФРызгЛђЗжзгНазіХфКЯЮяЁЃаЮГЩЬхЭЈГЃЪЧН№ЪєРызгКЭдзгЃЌвВгаЩйЪ§ЪЧЗЧН№ЪєдЊЫиЃЌР§ШчЃКCu2+,Ag+,Fe3+,Fe2+,Ni2+,BЂѓ,PЂѕ....
ХфЮЛЬхЭЈГЃЪЧЗЧН№ЪєЕФвѕРызгЛђЗжзгЃЌР§ШчЃКF-,Cl-,Br-,I-,OH-,CN-,H2O,NH3,CO.....
ХфЮЛдзгЁЊЁЊгыаЮГЩЬхГЩМќЕФдзгЃЛЕЅЛљХфЮЛЬхЁЊЁЊХфЮЛЬхжажЛгавЛИіХфЮЛдзгЃЛЖрЛљХфЮЛЬхЁЊЁЊОпгаСНИіЛђЖрИіХфЮЛдзгЕФХфЮЛЬхЁЃ
Р§ШчЃКввЖўАЗ(en)ЮЊСНИіХфЮЛдзгЃЛввЖўЫсИљЃЈВнЫсИљЃЉЮЊСНИіХфЮЛдзг)ЃЛввЖўАЗЫФввЫсИљ(МђГЦEDTA,ЛђY4-)(СљИіХфЮЛдзг)ЁЃ
ХфЮЛЪ§ЁЊЁЊХфЮЛдзгЪ§ЃКдкЕЅЛљХфЬхжаЃЌаЮГЩЬхЕФХфЮЛЪ§ЕШгкХфЮЛЪ§ЕФЪ§ФПЃЛдкЖрЛљХфЬхжаЃЌаЮГЩЬхЕФХфЮЛЪ§ЕШгкХфЮЛЬхЕФЪ§ФПгыЛљЪ§ЕФГЫЛ§ЁЃ
Р§ШчЃК[Cu(en)2]2+ЃЌCu2+ЕФХфЮЛЪ§ЕШгк4ЁЃ
ДгШмвКжаЮіГіХфКЯЮяЪБЁЃХфРызггыДјгаЯрЗДЕчКЩЕФЦфЫћРызгНсКЯГЩбЮЃЌетРрбЮЪєгкХфбЮЁЃХфбЮЕФзщГЩПЩвдЛЎЗжГЩФкВуКЭЭтВуЁЃХфРызгЪєгкФкВуЃЌХфРызгвдЭтЕФЦфЫћРызгЪєгкЭтВуЁЃЭтВуРызгЫљДјЕчКЩзмЪ§ЕШгкХфРызгЕФЕчКЩЪ§ЁЃ
ХфКЯЮяЕФЛЏбЇЪНКЭУќУћддђШчЯТЃК
ХфЫсЃКЁСЁСЁСЫсЃЛХфМюЃКЧтбѕЛЏЁСЁСЁСЃЛХфбЮЃКЯШвѕРызгКѓбєРызгЃЌМђЕЅЫсИљМгЁАЛЏЁАзжЁЃИДдгЫсИљМгЁАЫсЁБзжЁЃ
ФкВуУќУћЫГађЃКХфЮЛЪ§ЁЂХфЮЛУћГЦЁЂЁАКЯЁБЁЂаЮГЩЬхУћГЦЃЈбѕЛЏжЕЃЉЁЃХфЮЛЪ§ФПгУБЖЪ§ДЪЭЗЖўЁЂШ§ЁЂЫФЕШЪ§зжБэЪОЃЈХфЮЛЪ§ЮЊвЛЪБЪЁТдЃЉЃЌВЛЭЌХфЬхУћГЦжЎМфвдЁА."ЗжПЊЃЌдкзюКѓвЛИіХфЬхУћГЦжЎКѓзКвдЁАКЯЁБзжЁЃаЮГЩЬхЕФбѕЛЏжЕгУДјРЈКХЕФТоТэЪ§зжБэЪОЃЈбѕЛЏжЕЮЊ0ЪБЪЁТдЃЉЁЃ
ХфЬхДЮађЃКЯШРызгКѓЗжзгЁЃР§ШчЃКK[PtCl3NH3]УќУћЮЊШ§ТШЁЄАБКЯВЌ(Ђђ)ЫсМиЃЛ
ЭЌЪЧРызгЛђЭЌЪЧЗжзгЪБЃЌАДХфЮЛдзгЗћКХЕФгЂЮФзжФИЫГађХХСаЃЌР§ШчЃК[Co(NH3)5H2O]Cl3УќУћЮЊТШЛЏЮхАБЁЄЫЎКЯюм(Ђѓ)ЃЛ
ХфЮЛдзгЯрЭЌЃЌЩйдзгдкЯШЃЌХфЮЛдзгЯрЭЌЃЌЧвХфЬхжаКЌдзгЪ§ФПгжЯрЭЌЃЌАДЗЧХфЮЛдзгЕФдЊЫиЗћКХгЂЮФзжФИЫГађХХСаЃЌР§ШчЃК[PtNH2NO2(NH3)2]УќУћЮЊАБЛљЁЄЯѕЛљЁЄЖўАБКЯВЌ(ЂђЃЉЃЛ
ЯШЮоЛњКѓгаЛњЃЌР§ШчЃКK[PtCl3(C2H4)]УќУћЮЊШ§ТШЁЄввЯЉКЯВЌ(Ђђ)ЫсМиЁЃ
ПЮЬУУќУћСЗЯАЃКH2[PtCl6]ЃЛ[Cu(NH3)4](OH)2ЃЛ[Zn(OH)(H2O)3]NO3ЃЛ[Fe(CO)5]ЃЛK3[Fe(NCS)6]ЃЛ[Co(NO2)3(NH3)3]ЃЛ[Ca(EDTA)]2-
3.ХфКЯЮяЕФЗжРр
�������� МђЕЅХфКЯЮяЃКвЛИіжааФЁЂУПИіХфЬхОљЮЊЕЅЛљХфЮЛЬхЁЃШчЃК[Fe(CN)6]4-ЃЌ[Co(NH3)5(H2O)]3+ЁЃ
�������� ђќКЯЮяЃКвЛИіжааФРызггыЖрЛљХфЬхГЩМќаЮГЩЛЗзДНсЙЙЕФХфКЯЮяЁЃШчЃК[Cu(en)2]2+ЁЂCaY2-ЁЃ
�������� ЖрКЫХфКЯЮяЃККЌСНИіЛђСНИівдЩЯЕФжааФРызгЁЃШчЃК[(H2O)4Fe(OH)2(H2O)4]4+ЁЃ
�������� єЪКЯЮяЃКCOЮЊХфЬхЁЃШчЃКFe(CO)5ЃЌNi(CO)4ЁЃ
�������� ЯЉЬўЛЏКЯЮяЃКХфЬхЪЧВЛБЅКЭЬўЁЃШчЃК[PdCl3(C2H4)]-ЁЃ
�������� ЖрЫсаЭХфКЯЮяЃКХфЬхЪЧЖрЫсИљЁЃШчЃК(NH4)3[P(Mo3O10]ЁЄ6H2OЁЃ
ХфРызгЛђХфКЯЮяЗжзгдкЫЎШмвКжаДцдкзХХфКЯЮяЕФНтРыЗДгІКЭЩњГЩЗДгІжЎМфЕФЦНКтЃЌетжжЦНКтГЦЮЊХфЮЛЦНКтЁЃХфбЮШмгкЫЎЭъШЋНтРыЃЌНтРыГіЕФХфРызгдкЫЎШмвКжаЕФНтРыЯёШѕЕчНтжЪвЛбљЃЌЪЧВПЗжЕиЗжВННтРыГіЦфзщГЩВПЗжЁЃР§ШчЃК
![]()
![]()
змНтРыЗДгІЃК
![]()
![]() ЁЊЁЊ[Ag(NH3)2]+ЕФЗжВННтРыГЃЪ§
ЁЊЁЊ[Ag(NH3)2]+ЕФЗжВННтРыГЃЪ§
![]() ЁЊЁЊ[Ag(NH3)2]+ЕФзмНтРыГЃЪ§ЃЌгжГЦЮЊХфКЯЮяЕФВЛЮШЖЈГЃЪ§
ЁЊЁЊ[Ag(NH3)2]+ЕФзмНтРыГЃЪ§ЃЌгжГЦЮЊХфКЯЮяЕФВЛЮШЖЈГЃЪ§
![]()
![]() гњДѓЃЌХфКЯЮягњвзНтРыЃЌгњВЛЮШЖЈЁЃ
гњДѓЃЌХфКЯЮягњвзНтРыЃЌгњВЛЮШЖЈЁЃ
ХфКЯЮяЕФЩњГЩЗДгІЪЧХфКЯЮяНтРыЗДгІЕФФцЗДгІЁЃ
![]()
![]()
змЩњГЩЗДгІЃК![]()
![]() ЁЊЁЊ[Ag(NH3)2]+ЕФЗжВНЩњГЩГЃЪ§
ЁЊЁЊ[Ag(NH3)2]+ЕФЗжВНЩњГЩГЃЪ§
![]() ЁЊЁЊ[Ag(NH3)2]+ЕФзмЩњГЩГЃЪ§ЃЌгжГЦЮЊХфКЯЮяЕФЮШЖЈГЃЪ§ЛђРлЛ§ЮШЖЈГЃЪ§ЁЃ
ЁЊЁЊ[Ag(NH3)2]+ЕФзмЩњГЩГЃЪ§ЃЌгжГЦЮЊХфКЯЮяЕФЮШЖЈГЃЪ§ЛђРлЛ§ЮШЖЈГЃЪ§ЁЃ
![]()
![]() гњДѓЃЌХфКЯЮягњЮШЖЈЃЌВЛвзНтРыЁЃ
гњДѓЃЌХфКЯЮягњЮШЖЈЃЌВЛвзНтРыЁЃ
ПЩвдЭЦЕУЫќУЧЕФЙиЯЕЮЊЃК
![]()
![]()
![]()
ЧызЂвтЃКвЛАуЫЕРДЃЌХфКЯЮяЕФКЏМЖЮШЖЈГЃЪ§ЫцзХХфЮЛЪ§ЕФдіДѓЖјМѕаЁЃЌМД![]() ЕЋИїМЖЮШЖЈГЃЪ§жЎМфгаЪБВЛЪЧЯрВюЬЋДѓЃЌНјааЦНКтМЦЫуЪБЃЌШєРлЛ§ЮШЖЈГЃЪ§КмДѓЃЌЖјЧвХфЬхдкШмвКжаЕФХЈЖШвВНЯДѓЪБЃЌПЩзїНќЫЦМЦЫуЁЃ
ЕЋИїМЖЮШЖЈГЃЪ§жЎМфгаЪБВЛЪЧЯрВюЬЋДѓЃЌНјааЦНКтМЦЫуЪБЃЌШєРлЛ§ЮШЖЈГЃЪ§КмДѓЃЌЖјЧвХфЬхдкШмвКжаЕФХЈЖШвВНЯДѓЪБЃЌПЩзїНќЫЦМЦЫуЁЃ
ХфЬхШЁДњЗДгІЃКвЛжжХфЬхШЁДњСЫХфРызгФкВуЕФСэвЛжжХфЬхЃЌЩњГЩаТЕФХфКЯЮяЕФЗДгІГЦЮЊХфЬхШЁДњЗДгІЁЃЩњГЩЕФХфКЯЮяЕФЦНКтГЃЪ§дНДѓЃЌШЁДњЫљгУЕФХфКЯМСЕФХЈЖШдНДѓЃЌШЁДњЗДгІдНЭъШЋЁЃгаЪБЃЌХфЬхШЁДњЗДгІЗЂЩњЪБЃЌАщЫцзХШмвКЕФpHЕФИФБфЁЃ
ЙигкХфКЯЮяЮШЖЈадЕФНјвЛВНЬжТлЃЌИљОнИјЁЂЮќЕчзгФмСІЕФДѓаЁЃЌЮвУЧНЋЫсЁЂМюЗжЮЊгВЫсЁЂШэЫсКЭгВМюЁЂШэМюЕШЁЃ
ЫљЮНгВМюЃКЕчИКадДѓЃЈЮќв§ЕчзгФмСІЧПЃЉЁЂАыОЖаЁЃЌФббѕЛЏЃЈВЛвзЪЇШЅЕчзгЃЉЃЌВЛвзБфаЮЃЈФбБЛМЋЛЏЃЉЕФдзгЃЌвдетРрдзгЮЊХфЮЛдзгЕФМюЃЌГЦЮЊгВМюЃЌШчЃКNЃЌOЃЌFЕШЁЃ
ШэМюЃКЕчИКадаЁЃЈЮќв§ЕчзгФмСІШѕЃЉЃЌАыОЖНЯДѓЃЌвзБЛбѕЛЏЃЈвзЪЇШЅЕчзгЃЉЃЌШнвзБфаЮЃЈвзБЛМЋЛЏЃЉЕФдзг,ШчЃКI-ЃЌSCN-ЃЌS2-ЕШЃЌвдетРрдзгЮЊХфЮЛдзгЕФМюГЦЮЊШэМюЁЃ
НЛНчМюЃКНчгкгВМюКЭШэМюжЎМфЕФМюНазіНЛНчМюЁЃ
гВЫсЃКЕчКЩЪ§НЯДѓЁЂАыОЖНЯаЁЁЂЭтВуЕчзгБЛдзгКЫЪјИПЕУНЯНєЃЌвђЖјВЛШнвзБфаЮ(МЋЛЏТЪаЁЃЉЕФбєРызгЃЌШчЃКAl3+ЃЌFe3+ЃЌH+ЕШЁЃ
ШэЫсЃКЕчКЩЪ§НЯЩйЃЌАыОЖНЯДѓЃЌЭтВуЕчзгБЛдзгКЫЪјИПЕУБШНЯЧсЫЩвђЖјШнвзБфаЮЕФбєРызгЃЌШчЃКCu+ЃЌAg+ЃЌCd2+ЃЌHg2+ЕШЁЃ
НЛНчЫсЃКНчгкгВЫсКЭШэЫсжЎМфЕФЫсГЦЮЊНЛНчЫсЁЃ
LewisЫсМюИљОнгВШэЫсМюИХФюЗжРрКѓЃЌДѓСПЪЕбщЪТЪЕжЄУїЃКЁАгВЫсгыгВМюНсКЯЃЌШэЫсгыШэМюНсКЯЃЌГЃГЃаЮГЩЮШЖЈЕФХфКЯЮяЃЌМђГЦЮЊЁАгВЧзгВЃЌШэЧзШэЁБЁЃетвЛЙцТЩНазігВШэЫсМюддђЃЌГЦЮЊHSABддђЁЃ
зЂвтЃКгВШэЫсМюддђЛљБОЩЯЪЧОбщЕФЃЌБШНЯДжВкЃЌВЂВЛФмЗћКЯЫљгаЪЕМЪЧщПіЁЃгЩгкХфКЯЮяЕФГЩМќЧщПіБШНЯИДдгЃЌШЫУЧЖдШэгВЫсМюЕФбаОПЩаВЛЙЛЩюШыЃЌФПЧАЛЙВЛФмМђЕЅЕигУЁАгВЧзгВЃЌШэЧзШэЁБРДШЋУцВћЪіХфКЯЕФЮШЖЈадЁЃ
ФЧУДЃЌїЁКЯЮяЕФЮШЖЈадШчКЮЧѓЫуФиЃП
Бэ5-12вЛаЉђќКЯЮяКЭМђЕЅХфКЯЮяЕФЮШЖЈГЃЪ§ЃЌЖдБШБэжаЪ§ОнПЩжЊЃКЭЌвЛжжН№ЪєРызгЕФїЁКЯЮяЭљЭљБШОпгаХфЮЛдзгЕФХфЮЛЪ§ЕФМђЕЅХфКЯЮяЮШЖЈЃЈ![]() ДѓЃЉЃЌетжжЯжЯѓНазіїЁКЯаЇгІЁЃЭЈГЃЃЌїЁКЯХфЬхгыжааФРызгїЁКЯЖјГЩЮхдзгЛЗЛђСљдзгЛЗИќЮШЖЈаЉЃЌЧввЛИіїЁКЯХфЬхЬсЙЉЕФХфЮЛдзгдНЖрЃЌХХГЩЕФЮхдзгЛЗЛђСљдзгЛЗЕФЪ§вВдНЖрЃЌїЁКЯЮядНЮШЖЈЁЃ
ДѓЃЉЃЌетжжЯжЯѓНазіїЁКЯаЇгІЁЃЭЈГЃЃЌїЁКЯХфЬхгыжааФРызгїЁКЯЖјГЩЮхдзгЛЗЛђСљдзгЛЗИќЮШЖЈаЉЃЌЧввЛИіїЁКЯХфЬхЬсЙЉЕФХфЮЛдзгдНЖрЃЌХХГЩЕФЮхдзгЛЗЛђСљдзгЛЗЕФЪ§вВдНЖрЃЌїЁКЯЮядНЮШЖЈЁЃ
ЕкЪЎеТ МюН№ЪєКЭМюЭСН№ЪєдЊЫи
МюН№ЪєдЊЫидзгЕФМлЕчзгВуНсЙЙЮЊns1ЁЃвђДЫЃЌМюН№ЪєдЊЫижЛга+1бѕЛЏЬЌЁЃМюН№ЪєдзгзюЭтВужЛгавЛИіЕчзгЃЌДЮЭтВуЮЊ8Ечзг(LiЮЊ2Ечзг)ЃЌЖдКЫЕчКЩЕФЦСБЮаЇгІНЯЧПЃЌЫљвдетвЛИіМлЕчзгРыКЫаЃдЖЃЌЬиБ№ШнвзЪЇШЅЃЌвђДЫЃЌИїжмЦкдЊЫиЕФЕквЛЕчРыФмвдМюН№ЪєЮЊзюЕЭЁЃгыЭЌжмЦкЕФдЊЫиБШНЯЃЌМюН№ЪєдзгЬхЛ§зюДѓЃЌжЛгавЛИіГЩМќЕчзгЃЌдкЙЬЬхжадзгМфЕФв§СІНЯаЁЃЌЫљвдЫќУЧЕФШлЕуЁЂЗаЕуЁЂгВЖШЁЂЩ§ЛЊШШЖМКмЕЭЃЌВЂЫцзХLi-Na-K-Rb-CsЕФЫГађЖјЯТНЕЁЃЫцзХдзгСПЕФдіМг(МДдзгАыОЖдіМг)ЃЌЕчРыФмКЭЕчИКадвВвРДЮНЕЕЭЁЃ
МюН№ЪєаджЪЕФБфЛЏвЛАуКмгаЙцТЩЃЌЕЋгЩгкяЎдзгзюаЁЃЌЫљвдгааЉаджЪБэЯжЬиЪтЁЃЪТЪЕЩЯЃЌГ§СЫЫќУЧЕФбѕЛЏЬЌвдЭтЃЌяЎМАЦфЛЏКЯЮяЕФаджЪгыБОзхЦфЫќМюН№ЪєВюБ№НЯДѓЃЌЖјгыжмЦкБэжаяЎЕФгвЯТНЧдЊЫиУОгаКмЖрЯрЫЦжЎДІЁЃ
МюН№ЪєдЊЫидкЛЏКЯЪБЃЌЖрвдаЮГЩРызгМќЮЊЬиеїЃЌЕЋдкФГаЉЧщПіЯТвВЯдЙВМладЁЃЦјЬЌЫЋдзгЗжзгЃЌШчNa2ЁЂCs2ЕШОЭЪЧвдЙВМлМќНсКЯЕФЁЃМюН№ЪєдЊЫиаЮГЩЛЏКЯЮяЪБЃЌяЎЕФЙВМлЧуЯђзюДѓЃЌяЄзюаЁЁЃ
гыМюН№ЪєдЊЫиБШНЯЃЌМюЭСН№ЪєзюЭтВуга2ИіsЕчзгЁЃДЮЭтВуЕчзгЪ§ФПКЭХХСагыЯрСкЕФМюН№ЪєдЊЫиЪЧЯрЭЌЕФЁЃгЩгкКЫЕчКЩЯргІдіМгСЫвЛИіЕЅЮЛЃЌЖдЕчзгЕФв§СІвЊЧПвЛаЉЃЌЫљвдМюЭСН№ЪєЕФдзгАыОЖБШЯрСкЕФМюН№ЪєвЊаЁаЉЃЌЕчРыФмвЊДѓаЉЃЌНЯФбЪЇШЅЕквЛИіМлЕчзгЁЃЪЇШЅЕкЖўИіМлЕчзгЕФЕчРыФмдМЮЊЕквЛЕчРыФмЕФвЛБЖЁЃДгБэУцЩЯПДМюЭСН№ЪєвЊЪЇШЅСНИіЕчзгЖјаЮГЩЖўМле§РызгЫЦКѕКмРЇФбЃЌЪЕМЪЩЯЩњГЩЛЏКЯЮяЪБЫљЪЭЗХЕФОЇИёФмзувдЪЙЫќУЧЪЇШЅЕкЖўИіЕчзгЁЃЫќУЧЕФЕкШ§ЕчРыФмдМЮЊЕкЖўЕчРыФмЕФ4ЁЊ8БЖЃЌвЊЪЇШЅЕкШ§ИіЕчзгКмРЇФбЃЌвђДЫЃЌЫќУЧЕФжївЊбѕЛЏЪ§ЪЧ+2ЖјВЛЪЧ+1КЭ+3ЁЃгЩгкЩЯЪідвђЃЌЫљвдМюЭСН№ЪєЕФН№ЪєЛюЦУадВЛШчМюН№ЪєЁЃБШНЯЫќУЧЕФБъзМЕчМЋЕчЪЦЪ§жЕЃЌвВПЩвдЕУЕНЭЌбљЕФНсТлЁЃдкетСНзхдЊЫижаЃЌЫќУЧЕФдСЫАыОЖКЭКЫЕчКЩЖМгЩЩЯЖјЯТж№НЅдіДѓЃЌдкетРяЃЌдзгАыОЖЕФгАЯьЪЧжївЊЕФЃЌКЫЖдЭтВуЕчзгЕФв§СІж№НЅМѕШѕЃЌЪЇШЅЕчзгЕФЧуЯђж№НЅдіДѓЃЌЫљвдЫќУЧЕФН№ЪєЛюЦУадгЩЩЯЖјЯТж№НЅдіЧПЁЃ
МюН№ЪєКЭМюЭСН№ЪєЭХЬхОљЮЊН№ЪєОЇИёЃЌМюЭСН№ЪєгЩгкКЫЭтга2ИігааЇГЩМќЕчзгЃЌдгкМфОрРыНЯаЁЃЌН№ЪєМќЧПЖШНЯДѓЃЌвђДЫЃЌЫќУЧЕФШлЕуЁЂЗаЕуКЭгВЖШОљНЯМюН№ЪєИпЃЌЕМЕчадШДЕЭгкМюН№ЪєЁЃМюЭСН№ЪєЕФЮяРэаджЪБфЛЏВЛШчМюН№ЪєФЧУДгаЙцТЩЃЌетЪЧгЩгкМюЭСН№ЪєОЇИёРраЭВЛЪЧЭъШЋЯрЭЌЕФдЕЙЪЁЃМюН№ЪєНдЮЊЬхСЂЗНОЇИёЃЌМюЭСН№ЪєжаЃЌBeЁЂMgЮЊСљЗНОЇИёЃЌCaЁЂSrЮЊУцаФСЂЗНОЇИёЃЌBaЮЊЬхСЂЗНОЇИёЁЃ
етСНзхдЊЫиЕФРызгИїгаВЛЭЌЕФЮЖЕРЬиеїЃЌШчLi+РызгЮЖЬ№ЃЛK+ЁЂNa+РызгЮЖЯЬЃЛBa+РызгЮЖПрЁЃ
Li+РызгЕФМЋЛЏСІЪЧМюН№ЪєжазюЧПЕФЃЌЫќЕФШмМСЛЏзїгУКЭаЮГЩЙВМлЕФЧїЪЦвьГЃЕФДѓЃЌгаШЫЬсГігаЁАяЎМќЁБЕФДцдкЃЌРрЫЦгкЧтМќЃЌШчHЁЊFЁЄЁЄЁЄLiЁЊFКЭЃЈLiF2ЃЉ2ЁЃ
МюН№ЪєКЭМюЭСН№ЪєЕФживЊЮяРэаджЪЃКМюН№ЪєКЭМюЭСН№ЪєЕЅжЪГ§юыГЪИжЛвЩЋЭтЃЌЦфЫќЖМОпгавјАзЩЋЙтдѓЁЃМюН№ЪєОпгаУмЖШаЁЁЂгВЖШаЁЃЌШлЕуЕЭЁЂЕМЕчадЧПЕФЬиЕуЃЌЪЧЕфаЭЕФЧсН№ЪєЁЃМюЭСН№ЪєЕФУмЖШЃЌШлЕуКЭЗаЕудђНЯМюН№ЪєЮЊИпЁЃ
LiЁЂNaЁЂKЖМБШЫЎЧсЃЌяЎЪЧЙЬЬхЕЅжЪжазюЧсЕФЃЌЫќЕФУмЖШдМЮЊЫЎЕФвЛАыЁЃМюЭСН№ЪєЕФУмЖШЩдДѓаЉЃЌЕЋБЕЕФУмЖШБШГЃМћН№ЪєШчCuЁЂZnЁЂFeЛЙаЁКмЖрЁЃIAЁЂIIAзхН№ЪєЕЅжЪжЎЫљвдБШНЯЧсЃЌЪЧвђЮЊЫќУЧдкЭЌвЛжмЦкРяБШЯргІЕФЦфЫќдЊЫидзгСПНЯаЁЃЌЖјдзгАыОЖНЯДѓЕФдЕЙЪЁЃ
гЩгкМюН№ЪєЕФгВЖШаЁЃЌЫљвдФЦЁЂМиЖМПЩвдгУЕЖЧаИюЁЃЧаИюКѓЕФаТЯЪБэУцПЩвдПДЕНвјАзЩЋЕФН№ЪєЙтдѓЃЌНгДЅПеЦјвдКѓЃЌгЩгкЩњГЩбѕЛЏЮяЁЂЕЊЛЏЮяКЭЬМЫсбЮЕФЭтПЧЃЌбеЩЋБфАЕЁЃМюН№ЪєОпгаСМКУЕФЕМЕчадЁЃМюН№Ъє(ЬиБ№ЪЧМиЁЂяЈЁЂяЄ)дкЙтеежЎЯТЃЌФмЙЛЗХГіЕчзгЃЌЖдЙтЬиБ№СщУєЕФЪЧяЄЃЌЪЧЙтЕчГиЕФСМКУВФСЯЁЃяЈЁЂяЄПЩгУгкжЦдьзюзМШЗЕФМЦЪБЦїЁЊЁЊяЈЁЂяЄдзгжгЁЃ1967Фъе§ЪНЙцЖЈгУяЄдзгжгЫљЖЈЕФУыЮЊаТЕФЙњМЪЪБМфЕЅЮЛЁЃ
МюН№ЪєдкГЃЮТЯТФмаЮГЩвКЬЌКЯН№(77.2% KКЭ22.8% NaЃЌШлЕу260.7 K)КЭФЦЙЏЦы(ШлЕу236.2 K)ЃЌЧАепгЩгкОпгаНЯИпЕФБШШШКЭНЯПэЕФвКЛЏЗЖЮЇЖјБЛгУзїКЫЗДгІЖбЕФРфШДМСЃЌКѓепгЩгкОпгаЛККЭЕФЛЙдадЖјГЃдкгаЛњКЯГЩжагУзїЛЙдМСЁЃФЦдкЪЕбщЪвжаГЃгУРДГ§ШЅВаСєдкИїжжгаЛњШмМСжаЕФЮЂСПЫЎЗжЁЃ
яЎЕФгУЭОгњРДгњЙуЗКЃЌШчяЎКЭяЎКЯН№ЪЧвЛжжРэЯыЕФИпФмШМСЯЁЃяЎЕчГиЪЧвЛжжИпФмЕчГиЁЃ
МюЭСН№ЪєжаЪЕМЪгУЭОНЯДѓЕФЪЧУОЁЃжївЊгУРДжЦдьКЯН№ЁЃюызїЮЊаТаЫВФСЯШевцБЛжиЪгЁЃ
етСНзхдЊЫижагаМИжждЊЫидкЩњЮяНчгаживЊзїгУЁЃФЦКЭМиЪЧЩњЮяБиашЕФживЊдЊЫиЁЃУОЖдгкЫљгагаЛњНчЖМЪЧБиашЕФЁЃ
МюН№ЪєКЭМюЭСН№ЪєЕФживЊЛЏбЇаджЪЃКМюН№ЪєФЦЁЂМиЁЂИЦЁЂУОЗжБ№гыЫЎЗДгІЁЃН№ЪєФЦгыЫЎЗДгІОчСвЃЌВЂЗХГіH2ЁЃЗДгІЗХГіЕФШШЪЙФЦШлЛЏГЩаЁЧђЁЃМигыЫЎЕФЗДгІИќМЄСвЃЌВЂЗЂЩњШМЩеЃЌяЈЁЂяЄгыЫЎОчСвЗДгІВЂЗЂЩњБЌеЈЁЃ
МюЭСН№ЪєвВПЩвдгыЫЎЗДгІЁЃюыФмгыЫЎеєЦјЗДгІЃЌУОФмНЋШШЫЎЗжНтЃЌЖјИЦЁЂяШЁЂБЕгыРфЫЎОЭФмБШНЯОчСвЕиНјааЗДгІЁЃ
гЩДЫПЩжЊМюН№ЪєКЭМюЭСН№ЪєОљЮЊЛюЦУН№ЪєЃЌЖМЪЧЧПЛЙдМСЃЛдкЭЌвЛзхжаЃЌН№ЪєЕФЛюЦУадгЩЩЯЖјЯТж№НЅдіЧПЃЌдкЭЌвЛжмЦкжаДгзѓЕНгвН№ЪєЛюЦУадж№НЅМѕШѕЁЃ
ИљОнБъзМЕчМЋЕчЪЦЃЌяЎЕФЛюЦУадгІБШяЄИќДѓЃЌЕЋЪЕМЪЩЯгыЫЎЗДгІЛЙВЛШчФЦОчСвЁЃетЪЧвђЮЊ(1)яЎЕФШлЕуНЯИпЃЌЗДгІЪБВњЩњЕФШШСПВЛзувдЪЙЫќШлЛЏЃЌЖјФЦгыЫЎЗДгІЪБЗХГіЕФШШПЩвдЪЙФЦШлЛЏЃЌвђЖјЙЬЬхяЎгыЫЎНгДЅЕФЛњЛсВЛШчвКЬЌФЦЃЛ(2)ЗДгІВњЮяLiOHЕФШмНтЖШНЯаЁЃЌЫќИВИЧдкяЎЕФБэУцЃЌзшАЗДгІЕФНјааЁЃ
ЩЯЪіМюН№ЪєКЭМюЭСН№ЪєЕФЛюЦУадМАЦфБфЛЏЙцТЩЃЌЛЙБэЯждкЫќУЧдкПеЦјжаЖМШнвзКЭбѕЛЏКЯЁЃМюН№ЪєдкЪвЮТЯТФмбИЫйЕигыПеЦјжаЕФбѕЗДгІЃЌЫљвдМюН№ЪєдкПеЦјжаЗХжУвЛЖЮЪБЃЌН№ЪєБэУцОЭЩњГЩвЛВубѕЛЏЮяЃЌдкяЎЕФБэУцЩЯГ§ЩњГЩбѕЛЏЮяЭтЛЙгаЕЊЛЏЮяЁЃФЦЁЂМидкПеЦјжаЩдЮЂМгШШОЭШМЩеЦ№РДЃЌЖјяЈКЭяЄдкПеЮТЯТгіПеЦјОЭСЂМДШМЩеЁЃ
4Li+O2ЃН2Li2O
6Li+N2ЃН2Li3N
4Na+O2ЃН2Na2O
ЫќУЧЕФбѕЛЏЮядкПеЦјжавзЮќЪеЖўбѕЛЏЬМаЮГЩЬМЫсбЮЃК
Na2O+CO2=Na2CO3
вђДЫМюН№ЪєгІДцЗХдкУКгЭжаЃЌвђяЎЕФУмЖШзюаЁЃЌПЩвдИЁдкУКгЭЩЯЃЌЫљвдНЋЦфНўдквКЬхЪЏРЏЛђЗтДцдкЙЬЬхЪЏРАжаЁЃ
МюЭСН№ЪєЛюЦУадТдВюЃЌЪвЮТЯТетаЉН№ЪєБэУцЛКТ§ЩњГЩбѕЛЏФЄЁЃЫќУЧдкПеЦјжаМгШШВХЯджјЗЂЩњЗДгІЃЌГ§ЩњГЩбѕЛЏЮяЭтЃЌЛЙгаЕЊЛЏЮяЩњГЩЁЃ
3Ca+N2=Ca3N2
вђДЫдкН№ЪєШлСЖжаГЃгУLiЁЂCaЕШзїЮЊГ§ЦјМСЃЌГ§жЇШмНтдкШлШкН№ЪєжаЕФЕЊЦјКЭбѕЦјЁЃ
дкИпЮТЪБМюН№ЪєКЭМюЭСН№ЪєЛЙФмЖсШЁФГаЉбѕЛЏЮяжаЕФбѕЃЌШчУОПЩЪЙSiO2ЕФЙшЛЙдГЩЕЅжЪSiЃЌЛђЖсШЁТШЛЏЮяжаЕФТШЃЌШчН№ЪєФЦПЩвдДгT1Cl4жажУЛЛГіН№ЪєюбЁЃ
SiO2+2MgЃНSi+2MgO
T1C14+4NaЃНTi+4NaCl
МюН№ЪєзюгааЫШЄЕФЯИжТжЎвЛЪЧЫќУЧдквКАБжаБэЯжЕФаджЪЁЃМюН№ЪєЕФвКАБЯЁШмвКГЪРЖЩЋЃЌЫцзХМюН№ЪєШмНтСНЕФдіМгЃЌШмвКЕФбеЩЋБфЩюЁЃЕБДЫШмвКжаФЦЕФХЈЖШГЌЙ§1mol/LвдКѓЃЌОЭдкдРДЩюРЖЩЋШмвКжЎЩЯГіЯжвЛИіЧрЭЩЋЕФаТЯрЁЃдйЬэМгМюН№ЪєЃЌШмвКОЭгЩРЖЩЋБфЮЊЧрЭЩЋЁЃШчНЋШмвКеєЗЂЃЌгжПЩвджиаТЕУМюН№ЪєЁЃ
ИљОнбаОПШЯЮЊЃКдкМюН№ЪєЕФЯЁАБШмвКжаМюН№ЪєРыНтЩњГЩМюН№Ъєе§РызгКЭШмМСКЯЕчзгЁЃвђЮЊРыНтЩњГЩАБКЯбєРызгКЭАБКЯЕчзгЃЌЫљвдШмвКгаЕМЕчадЁЃДЫШмвКОпгаИпЕМЕчаджївЊЪЧгЩгкгаШмМСКЯЕчзгДцдкЁЃШмвКжавђКЌгаДѓСПШмМСКЯЕчзгЃЌвђДЫЪЧЫГДХадЕФЁЃКлСПдгжЪШчЙ§ЖЩН№ЪєЕФбЮРрЁЂбѕЛЏЮяКЭЧтбѕЛЏЮяЕФДцдкЃЌвдМАЙтЛЏзїгУЖМФмДйНјШмвКжаЕФМюН№ЪєКЭвКАБжЎМфЗЂЩњЗДгІЖјЩњГЩАБЛљЛЏЮяЃК
Na+NH3ЃЈlЃЉ=NaNH2+1/2H2
ИЦЁЂяШЁЂБЕвВФмШмгквКАБЩњГЩКЭМюН№ЪєвКАБШмвКЯрЫЦЕФРЖЩЋШмвКЃЌгыФЦЯрБШЃЌЫќУЧШмЕУвЊТ§аЉЃЌСПвВЩйаЉЁЃМюН№ЪєвКАБШмвКжаЕФШмМСКЯЕчзгЪЧвЛжжКмЧПЕФЛЙдМСЁЃЫќУЧЙуЗКгІгУдкЮоЛњКЭгаЛњжЦБИжаЁЃ
SЧјдЊЫиЕФЛ№бцбцЩЋЗДгІЃКМюН№ЪєКЭИЦЁЂяШЁЂБЕЕФЛгЗЂадбЮдкЮоЩЋЛ№бцжазЦЩеЪБЃЌФмЪЙЛ№бцГЪЯжГівЛЖЈбеЩЋЁЃетНаЁАбцЩЋЗДгІЁБЁЃМюН№ЪєКЭИЦЁЂяШЁЂБЕЕФбЮЃЌдкзЦЩеЪБЮЊЪВУДФмВњЩњВЛЭЌЕФбеЩЋФиЃПвђЮЊЕБН№ЪєЛђЦфбЮдкЛ№бцЩЯзЦЩеЪБЃЌдзгБЛМЄЗЂЃЌЕчзгНгЪмСЫФмСПДгНЯЕЭЕФФмМЖЬјЕННЯИпФмМЖЃЌЕЋДІдкНЯИпФмМЖЕФЕчзгЪЧКмВЛЮШЖЈКмПьЬјЛиЕНЕЭФмМЖЃЌетЪБОЭНЋЖргрЕФФмСПвдЙтЕФаЮЪНЗХГіЁЃдзгЕФНсЙЙВЛЭЌЃЌОЭЗЂГіВЛЭЌВЈГЄЕФЙтЃЌЫљвдЙтЕФбеЩЋвВВЛЭЌЁЃМюН№ЪєКЭМюЭСН№ЪєЕШФмВњЩњПЩМћЙтЦзЃЌЖјЧвУПвЛжжН№ЪєдзгЕФЙтЦзЯпБШНЯМђЕЅЃЌЫљвдШнвзЙлВьЪЖБ№ЁЃРћгУбцЩЋЗДгІЃЌПЩвдИљОнЛ№бцЕФбеЩЋЖЈадЕФМјБ№етаЉдЊЫиЕФДцдкгыЗёЃЌЕЋвЛДЮжЛФмМјБ№вЛжжРызгЁЃЭЌЪБРћгУМюН№ЪєКЭИЦЁЂяШЁЂБЕбЮдкзЦЩеЪБВњЩњВЛЭЌбцЩЋЕФдРэЃЌПЩвджЦдьИїЩЋбцЛ№ЃЌР§ШчКьЩЋбцЛ№ЕФМђЕЅХфЗНЃК
жЪСПАйЗжБШKClO334%Sr(NO3)2ЬПЗл10%УОЗл4%ЫЩЯу7%
ТЬЩЋбцЛ№ХфЗНЃК
жЪСПАйЗжБШBa(ClO3)238%Ba(NO3)240%S22%
гЩгкМюН№ЪєКЭМюЭСН№ЪєЕФЛЏбЇаджЪКмЛюЦУЃЌЫљвдЫќУЧжЛФмвдЛЏКЯзДЬЌДцдкгкздШЛНчжаЁЃдкМюН№ЪєжаЃЌФЦКЭяЎдкЕиПЧжаЗжВМКмЙуЃЌСНепЕФЗсЖШЖМЮЊ2.5%ЁЃжївЊПѓЮягаФЦГЄЪЏNa[AlSi3O8]ЁЂКЭМиГЄЪЏK[A1Si3O8]ЃЌЙтТБЪЏKClЁЄMgCl2ЁЄ6H20МАУїЗЏЪЏK2SO4ЁЄA12(SO4)3ЁЄ24H2OЕШЁЃКЃЫЎжаТШЛЏФЦЕФКЌСПЮЊ2.7ЃЅЃЌжВЮяЛвжавВКЌгаМибЮЁЃяЎЕФживЊПѓЮяЮЊяЎЛдЪЏLi20ЁЄA12034SiO2ЃЌяЎЁЂяЈКЭяЄдкздШЛНчжаДЂСПНЯЩйЧвЗжЩЂЃЌБЛСаЮЊЯЃгаН№ЪєЁЃ
МюЭСН№ЪєГ§РиЭтдкздШЛНчаЁЗжВМвВКмЙуЗКЃЌУОГ§ЙтТБЪЏЭтЃЌЛЙгаАздЦЪЏCaCO3ЁЄMgCO3КЭСтУОПѓMgCO3ЕШЁЃюыЕФзюживЊПѓЮяЪЧТЬжљЪЏ3BeOЁЄAl2O3ЁЄ6SiO3ЁЃИЦЁЂяШЁЂБЕдкздШЛНчжаДцдкЕФжївЊаЮЪНЮЊФбШмЕФЬМЫсбЮКЭСђЫсбЮЃЌШчЗННтЪЏCaCO3ЁЂЬМЫсяШПѓSrCO3ЁЂЬМЫсБЕПѓЁЂЪЏИрCaSO4ЁЄ2H2OЁЂЬьЧрЪЏSrSO4бГдбББЪЏBaSO4ЕШЁЃКЃЫЎжаКЌгаДѓСПУОЕФТШЛЏЮяКЭСђЫсбЮЃЌ1971ФъЪРНчУОВњСПгавЛАывдЩЯЪЧвдКЃЫЎЮЊдСЯЩњВњЕФЁЃгЩгкМюН№ЪєКЭМюЭСН№ЪєЕФаджЪКмЛюЦУЃЌЫљвдвЛАуЖМгУЕчНтЫќУЧЕФШлШкЛЏКЯЮяЕФЗНЗЈжЦШЁЁЃФЦКЭяЎжївЊгУЕчНтШлШкЕФТШЛЏЮяжЦШЁЁЃ
ЕчНтШлШкТШЛЏФЦжЦН№ЪєФЦЃКжЦШЁН№ЪєФЦЕчНтВлЪОвтЭМШчЯТЁЃЕчНтВлЭтгаИжПЧЃЌФкГФФЭЛ№ВФСЯЁЃСНМЋгУИєЧНЗжПЊЁЃТШЦјДгбєМЋЧјЩЯВПЙмЕРХХГіЃЌФЦДгвѕМЋЧјГіПкСїГіЁЃ
ЕчНтгУЕФдСЯЪЧТШЛЏФЦКЭТШЛЏИЦЕФЛьКЯбЮЁЃШєжЛгУТШЛЏФЦНјааЕчНтЃЌВЛНіашвЊИпЮТЃЌЖјЧвЕчНтЮіГіЕФН№ЪєФЦвзЛгЗЂ(ТШЛЏФЦЕФШлЕуЮЊ1073KЃЌФЦЕФЗаЕуЮЊ1156K)ЃЌЛЙШнвзЗжЩЂдкШлШкбЮжаЃЌФбгкЗжРыГіРДЁЃМгШыТШЛЏИЦКѓЃЌвЛдђПЩНЕЕЭЕчНтжЪЕФШлЕу(ЛьКЯбЮЕФШлЕудМ873K)ЃЌЗРжЙФЦЕФЛгЗЂЃЌдйдђПЩМѕаЁН№ЪєФЦЕФЗжЩЂадЃЌвђШлШкЛьКЯЮяЕФУмЖШБШН№ЪєФЦДѓЃЌФЦвзИЁдкУцЩЯЁЃ
жЦШЁМюН№ЪєЕФЗНЗЈЛЙгаШШЛЙдЗЈЁЂН№ЪєжУЛЛЗЈКЭШШЗжНтЗЈЁЃ
ШШЛЙдЗЈЃКвЛАуВЩгУНЙЬПЛђЬМЛЏЮяЮЊЛЙдМС
Н№ЪєжУЛЛЗЈЃКМиЁЂяЈКЭФЦЫфШЛвВПЩвдгУЕчНтЗЈжЦШЁЃЌЕЋГЃгУЧПЛЙдадЕФН№ЪєШчNaЁЂCaЁЂMgЁЂBaЕШдкИпЮТКЭЕЭбЙЯТЛЙдЫќУЧТШЛЏЮяЕФЗНЗЈжЦШЁЁЃ
ЩЯУцМИИіЗДгІПДЦ№РДФЧЪЧНЯВЛЛюЦУЕФН№ЪєАбЛюЦУН№ЪєДгЦфбЮРржажУЛЛГіРДЃЌетЫЦКѕгыН№ЪєЕФБъзМЕчМЋЕчЪЦХХСаЕФН№ЪєЛюЖЏЫГађЯрУЌЖмЃЌЮвУЧвбОжЊЕРгУБъзМЕчМЋЕчЪЦзїЗДгІЗНЯђЕФХаЖЯБъзМЃЌжЛФмдкЫЎШмвКЕФЧщПіЯТгІгУЃЌЖјЩЯЪіЗДгІЖМЪЧдкИпЮТЯТНјааЕФЃЌЫљвдВЛФмгІгУЁЃНЋФЦеєЦјЭЈШыШлШкЕФKClжаЃЌПЩвдЕУЕНвЛжжФЦЁЊМиКЯН№ЁЃДгБэ17ЁЊ3ПЩжЊЃКФЦЕФЗаЕуЮЊ1155ЃЎ9KЃЌМиЮЊ1047.9KЃЌМидкИпЮТИќвзЛгЗЂЁЃдквЛИіЗжСѓЫўжаМгШШЁЃРћгУМидкИпЮТЪБЛгЗЂЖШДѓЖјДгКЯН№жаЗжРыГіРДЁЃСэЭтФЦКЭМиЕФЭЌРраЭЛЏКЯЮяЕФОЇИёФмЯрБШЃЌФЦБШМиИпЃЌвђЖјФЦЕФЛЏКЯЮяИќЮШЖЈЁЃ
МиЗаЕуЕЭвзЛгЗЂЃЌМивзШлгкШлШкKClжаФбЗжРыЃЌдкЕчНтЙ§ГЬжаВњЩњЕФKO2гыKЛсЗЂЩњБЌеЈ,ЫљвдвЛАуВЛгУШлШкбЮЕчНтЗЈжЦШЁМиЃЌжївЊгУН№ЪєжУЛЛЗЈЕШжЦШЁЁЃ
ШШЗжНтЗЈЃКМюН№ЪєЕФЛЏКЯЮяЃЌШчбЧЬњЧшЛЏЮяЃЌЧшЛЏЮяКЭЕўЕЊЛЏЮяЃЌМгШШФмБЛЗжНтГЩМюН№ЪєЁЃ
МюН№ЪєЕФЕўЕЊЛЏЮяНЯвзДПЛЏЃЌЖјЧвВЛвЛЗЂЩњБЌеЈЁЃетжжЗНЗЈЪЧОЋШЗЖЈСПжЦБИМюН№ЪєЕФРэЯыЗНЗЈЁЃяЎвђаЮГЩКмЮШЖЈЕФLi3NЃЌЙЪВЛФмгУетжжЗНЗЈжЦБИЁЃ
МюН№ЪєКЭМюЭСН№ЪєЕФЧтЛЏЮяЃКЛЏбЇЛюадКмИпЕФМюН№ЪєКЭМюЭСН№ЪєжаНЯЛюЦУЕФCaЁЂSrЁЂBaФмгыЧтдкИпЮТЯТжБНгЛЏКЯЃЌЩњГЩРызгаЭЧтЛЏЮяЃК
2M+H2ЃН2M+H-(MЃНМюШЋЪє)
M+H2ЃНM2+H2-(M=CaЁЂSrЁЂBa)
ЧтЛЏяЎдМдк998KЪБаЮГЩЃЌЧтЛЏФЦКЭЧтЛЏМидк573ЁЊ673KЪБЩњГЩЃЌЦфгрЧтЛЏЮядк723KЪБЩњГЩЕЋдкГЃбЙЯТЗДгІНјааЛКТ§ЁЃетаЉЧтЛЏЮяОљЮЊАзЩЋОЇЬхЃЌЕЋГЃвђЛьгаКлСПН№ЪєЖјЗЂЛвЁЃгЩгкМюН№ЪєКЭCaЁЂSrЁЂBaгыЧтЕФЕчИКадЯрВюНЯДѓЃЌЧтДгН№ЪєдзгЕФЭтВуЕчзгжаЖсЕУ1ИіЕчзгаЮГЩвѕРызгH-ЃЌетаЉЧтЛЏЮяЖМЪЧРызгОЇЬхЃЌЙЪГЦЮЊРызгаЭЧтЛЏЮяЃЌгжГЦЮЊбЮаЭЧтЛЏЮяЁЃЕчНтШлШкЕФбЮаЭЧтЛЏЮяЃЌдкбєМЋЩЯЗХГіЧтЦјЃЌжЄУїдкетРрЧтЛЏЮяжаЕФЧтЪЧДјИКЕчЕФзщЗжЁЃМюН№ЪєЧтЛЏЮяИіЕФH-РызгЕФАыОЖНщгкМюН№ЪєЗњБШЮяжаЕФF-РызгКЭТШЛЏЮяжаЕФC1-РызгжЎМфЃЌвђДЫЃЌМюН№ЪєЧтЛЏЮяЕФФГаЉаджЪРрЫЦгкЯргІЕФМюН№ЪєТБЛЏЮяЁЃ
МюН№ЪєЧтЛЏЮяжавдLiHзюЮШЖЈЁЃМгШШЕНШлЕу(961K)вВВЛЗжНтЁЃЦфЫќМюН№ЪєЧтЛЏЮяЮШЖЈадНЯВюЃЌМгШШЛЙВЛЕНШлЕуЃЌОЭЗжНтГЩН№ЪєКЭЧтЁЃ
ЫљгаМюН№ЪєЧтЛЏЮяЖМЪЧЧПЛЙдМСЁЃЙЬЬЌNaHдк673KЪБФмНЋTiCl4ЛЙдЮЊН№ЪєюбЃК
TiCl4+4NaHЃНTi+4NaCl+2H2
LiHКЭCaH2ЕШдкгаЛњКЯГЩжаГЃзїЮЊЛЙдМСЁЃдкЫЎШмвКH2ЃЏH-ЕчЖдЕФE0=225VЃЌПЩМћH-ЪЧзюЧПЕФЛЙдМСжЎвЛЃЌЫќУЧгіЕНКЌгаH+ЕФЮяжЪЃЌШчЫЎЃЌОЭбИЫйЗДгІЖјЗХГіЧтЃК
LiH+H2OЃНLiOH+H2
CaH2+2H2OЃНCa(OH)2+2H2
гЩЧтЛЏИЦгыЫЎЗДгІЖјФмЗХГіДѓСПЕФЧтЦјЃЌЫљвдГЃгУЫќзїЮЊвАЭтЗЂЩњЧтЦјЕФВФСЯЁЃ
МюН№ЪєгыбѕЛЏКЯПЩвдаЮГЩЖржжбѕЛЏЮяЃЌЦеЭЈбѕЛЏЮяM2OЃЌЙ§бѕЛЏЮяM2O2ЃЌГЌбѕЛЏЮяMO2КЭГєбѕЛЏЮяMO3ЁЃМюН№ЪєдкЙ§СПЕФПеЦјжаШМЩеЪБЃЌЩњГЩВЛЭЌРраЭЕФбѕЛЏЮяЃКШчяЎЩњГЩбѕЛЏяЎLi2OЃЌФЦЩњГЩЙ§бѕЛЏФЦЃЌЖјМиЁЂяЈЁЂяЄдђЩњГЩГЌбѕЛЏЮяЁЃМюЭСН№ЪєвЛАуЩњГЩЦеЭЈбѕЛЏЮяMOЃЌИЦЁЂяШЁЂБЕЛЙПЩвдаЮГЩЙ§бѕЛЏЮяКЭГЌбѕЛЏЮяЁЃ
МюН№ЪєКЭМюЭСН№ЪєЕФбѕЛЏЮяЃКдкПеЦјжаШМЩеЪБЃЌжЛгаяЎЩњГЩбѕЛЏяЎ(АзЩЋЙЬЬх)ЁЃОЁЙмдкШБбѕЕФПеЦјжаПЩвджЦЕУГ§яЎвдЭтЕФЦфЫќМюН№ЪєЦеЭЈбѕЛЏЮяЃЌЕЋетжжЬѕМўВЛвзПижЦЃЌЫљвдЦфЫќМюН№ЪєЕФбѕЮяM2OБиаыВЩгУМфНгЗНЗЈРДжЦБИЁЃР§ШчгУН№ЪєФЦЛЙдЙ§бѕЛЏФЦЃЌгУН№ЪєМиЛЙдЯѕЫсМиЃЌЗжБ№ПЩвджЦЕУбѕЛЏФЦ(АзЩЋЭХЬх)КЭбѕЛЏМи(ЕЛЦЩЋЙЬЬх)ЃК
Na2O2+2NaЃН2Na2O
2KN03+10KЃН6K2O+N2
Rb2OЮЊССЛЦЩЋЃЌCs2OЮЊГШКьЩЋЃЌбѕЛЏЮяЕФбеЩЋвРДЮМгЩюЁЃ
МюН№ЪєбѕЛЏЮяM2OгыЫЎЛЏКЯЖјЩњГЩЧтбѕЛЏЮяMOHЃК
M2O+H2O=2MOH
МюН№ЪєбѕЛЏЮягыЫЎЗДгІЕФГЬЖШЃЌДгLi2OЕНCs2OвРДЮМгЧПЁЃLi2OгыЫЎЗДгІКмТ§ЃЌЕЋRb2OКЭCs2OгыЫЎЗДгІЪБЛсЗЂЩњШМЩеЩѕжСБЌеЈЁЃ
МюЭСН№ЪєдкЪвЮТЛђМгШШЯТЃЌФмКЭбѕЦјжБНгЛЏКЯЖјЩњГЩбѕЛЏЮяMOЃЌБШПЩвдДгЫќУЧЕФЬМЫсбЮЛђЯѕЫсбЮМгШШЗжНтжЦЕУMOЃЌР§ШчЃК
CaCO3=CaO+CO2Ёќ
2Sr(NO3)2=2SrO+4NO2Ёќ+O2Ёќ
бѕЛЏИЦгыЫЎЗДгІЖјЩњГЩЪьЪЏЛвВЂЗХГіДѓСПЕФШШЃЌЪьЪЏЛвЙуЗКгІгУдкНЈжўЙЄвЕЩЯЁЃ
CaO+H2O=Ca(OH)2
МюЭСН№ЪєбѕЛЏЮяЕФЫЎКЯШШДгBeЕНBaвРДЮдіМгЁЃбѕЛЏИЦЕФетжжЫЎКЯФмСІЃЌГЃгУРДЮќЪеОЦОЋжаЕФЫЎЗжЁЃдкИпЮТЯТбѕЛЏИЦФмЭЌЫсадбѕЛЏЮяSiO2зїгУЃК
CaO+SiO2ЃНCaSiO3
CaOгыP2O5вВгаРрЫЦЗДгІЃЌетПЩгУдкСЖИжжаГ§ШЅдгжЪСзЁЃ
МюЭСН№ЪєЕЊЛЏЮяЖМЪЧАзЩЋЭХЬхЁЃГ§BeOЭтЃЌЖМЪЧТШЛЏФЦОЇИёЕФРызгаЭЛЏКЯЮяЁЃгЩгке§ЁЂИКРызгЖМЪЧДјгаСНИіЕчКЩЃЌЖјMЁЊOЕФОрРыгжНЯаЁЃЌЫљвдMOОпгаНЯДѓЕФОЇИёФмЃЌвђДЫЫќУЧЕФШлЕуКЭгВЖШЖМУЧЕБИпЁЃОЇИёжаРызгМфОрРывРДЮНЕЕЭЃЌШлЕуГ§BeOЭтвВЪЧвРДЮЯТНЕЁЃИљОнетжжЬиадЃЌBeOКЭMgOГЃгУРДжЦдьФЭЛ№ВФСЯКЭН№ЪєЬеДЩЁЃ
УмЖШЮЊ2.949ЁЄcm-3ЕФMgOЮЊАзЩЋЯИФЉЃЌГЦЧсжЪбѕЛЏУОЁЃУмЖШЮЊ3.589ЁЄcm-3ЕФMgOГЦжижЪбѕЛЏУОЁЃЫћУЧОљФбШмгкЫЎЃЌвзШмгкЫсКЭАБбЮШмвКЁЃбѕЛЏУОНўгкЫЎжаТ§Т§зЊБфЮЊЧтбѕЛЏУОЁЃ
МюН№ЪєКЭМюЭСН№ЪєЕФЙ§бѕЛЏЮяЃКЙ§бѕЛЏЮяM2O2жаКЌгаЙ§бѕЛЏРызгO22-Лђ[ЁЊOЁЊOЁЊ]2-ЁЃЦфЗжзгЙьЕРЪНШчЯТЃК
[KKЃЈІФ2SЃЉ2ЃЈІФ*2SЃЉ2ЃЈІФ2PЃЉ2ЃЈІа2PЃЉ4ЃЈІа*2PЃЉ4]
ГЩМќКЭЗДМќІаЙьЕРДѓжТЕжЯћЃЌгЩЬюГфІФ2PxЙьЕРЕФЕчзгаЮГЩвЛИіІФМќЃЌМќМЖЮЊ1ЁЃ
МюН№ЪєзюГЃМћЕФЙ§бѕЛЏЮяЪЧЙ§бѕЛЏФЦЃЌЪЕМЪгУЭОвВНЯДѓЁЃНЋФЦМгШШжСШлЛЏЃЌЭЈШывЛЖЈСПЕФГ§ШЅCO2ЕФИЩдяПеЦјЃЌЮЌГжЮТЖШдк453-473KжЎМфЃЌФЦМДБЛбѕЛЏЮЊNa2OЃЛНјЖјдіМгПеЦјСїСПВЂбИЫйЬсИпЮТЖШжС573-673KЃЌМДПЩжЦЕУNa2O(ЕЛЦЩЋЗлФЉ)ЃК
Na2O2гыЫЎЛђЯЁЫсЗДгІЖјВњЩњH2O2ЃЌH2O2СЂМДЗжНтЗХГібѕЦјЃК
Na2O2+2H2OЃНH2O2+2NaOH
Na2O2+H2SO4ЃНH2O2+Na2SO4
2H2O2ЃН2H2O+O2Ёќ
ЫљвдNa2O2ПЩгУзїбѕЛЏМСЁЂЦЏАзМСКЭбѕЦјЗЂЩњМСЁЃNa2O2гыCO2ЗДгІЃЌвВФмЗХГібѕЦјЃК
2Na2O2+2CO2ЃН2Na2CO3+O2
РћгУетвЛаджЪЃЌNa2O2дкЗРЖОУцОпЁЂИпПеЗЩааКЭЧБЭЇжагУзїCO2ЕФЮќЪеМСКЭЙЉбѕМСЁЃ
Й§бѕЛЏФЦдкМюадНщжЪжаЪЧвЛжжЧПбѕЛЏМСЃЌР§ШчдкМюадШмвКжаЃЌЫќПЩвдАбAs(III)бѕЛЏГЩAs(v)ЕФЛЏКЯЮяЃЌАбCr(III)бѕЛЏГЩCr(IV)ЕФЛЏКЯЮяЕШЁЃдкЗжЮіЛЏбЇжаЃЌГЃгУЫќРДбѕЛЏЗжНт(МюШл)ФГаЉПѓЮяЁЃР§ШчЃЌЫќФмНЋПѓЪЏжаСђЁЂУЬЁЂИѕЁЂЗАЁЂЮ§ЕШГЩЗжбѕЛЏГЩПЩШмЕФКЌбѕЫсбЮЃЌЖјздЪдбљжаЗжРыГіРДЃЌвђДЫГЃгУзїЗжНтПѓЪЏЕФШлМСЁЃР§Шч
Cr2O3+3Na2O2ЃН2Na2CrO4+Na2O
MnO2+Na2O2ЃНNa2MnO4
гЩгкNa2O2гаЧПМюадЃЌШлШкЪБВЛФмВЩгУДЩжЦЦїУѓЛђЪЏгЂЦїУѓЃЌвЫгУЬњЁЂФјЦїУѓЁЃгЩгкЫќУЧЧПбѕЛЏадЃЌШлШкЪБгіЕНУоЛЈЁЂЬПЗлЛђЧІЗлЛсЗЂЩњБЌеЈЃЌЪЙгУЪБгІЪЎЗжаЁаФЁЃ
МюЭСН№ЪєЕФЙ§бѕЛЏЮявдBaO2НЯЮЊживЊЁЃЙ§бѕЛЏБЕЛЙПЩзїЙЉбѕМСЁЂв§Л№МСЕШ
МюН№ЪєКЭМюЭСН№ЪєЕФГЌбѕЛЏЮяЃКМиЁЂяЈЁЂяЄдкЙ§СПЕФбѕЦјжаШМЩеМАЕУГЌбѕЛЏЮяMO2ЁЃKO2ЪЧГШЛЦЩЋЙЬЬхЃЌRbO2ЪЧЩюзиЩЋЙЬЬхЃЌCsO2ЪЧЩюЛЦЩЋЙЬЬхЁЃГЌбѕЛЏЮяжаКЌгаГЌбѕРызгO2-,ЦфНсЙЙЮЊЃК[OЁЄЁЄЁЄO]-
ЦфЗжзгЙьЕРЪНЮЊЃКO2-[KKЃЈІФ2SЃЉ2ЃЈІФ*2SЃЉ2ЃЈІФ2PЃЉ2ЃЈІа2PЃЉ4ЃЈІа*2PЃЉ3]
дкO2-жаЃЌга13ИіМлЕчзгЃЌЦфжаГЩМќКЭЗДМќЙьЕРДѓжТЕжЯћЃЌГЩМќЕФЃЈІФ2PЃЉ2ЙЙГЩвЛИіІФМќЃЌГЩМќЕФЃЈІа2PЃЉ2КЭЗДМќЕФЃЈІа*2PЃЉ1ЙЙГЩвЛИіШ§ЕчзгІаМќМќМЖЮЊЃКЃЈ2+2-1ЃЉ/2=1![]()
вђГЌбѕРызгO2-гавЛИіФЉГЩЖдЕФЕчзгЃЌЙЪЫќОпгаЫГДХадЃЌВЂГЪЯжГібеЩЋЁЃгЩгкO2-ЕФМќМЖБШO2аЁЃЌЫљвдЮШЖЈадБШO2ВюЁЃЪЕМЪЩЯГЌбѕЛЏЮяЪЧЧПбѕЛЏМСЃЌгыЫЎОчСвЕиЗДгІЃК
2MO2+2H2OЃНO2+H2O2+2MOH
вВФмКЭCO2ЗДгІЗХГібѕЦјЃК
4MO2+2CO2ЃН2M2CO3+3O2
ЙЪЫќУЧвВФмГ§ШЅCO2КЭдйЩњO2ЃЌвВПЩгУгкМБОШЦїжаКЭЧБЫЎЁЂЕЧЩНЕШЗНУцЁЃ
ДЫЭтГєбѕКЭKЁЂRbЁЂCsЕФЧтбѕЛЏЮязїгУЃЌПЩвджЦЕУГєбѕЛЏЮяЃЌР§ШчЃК
3KOH(s)+2O3(g)ЃН2KO3(s)+KOHЁЄH2O(s)+O2(g)
НЋKO3гУвКАБжиНсЦЗЃЌПЩЕУЕННлКьЩЋЕФKO3ОЇЬхЃЌЫќЛКТ§ЕиЗжНтГЩKO2КЭO2ЁЃ
МюН№ЪєКЭМюЭСН№ЪєЕФЧтбѕЛЏЮяЃКМюН№ЪєЕФЧтбѕЛЏЮяЖдЯЫЮЌКЭЦЄЗєгаЧПСвЕФИЏЪДзїгУЃЌЫљвдГЦЫќУЧЮЊПСадМюЁЃЧтбѕЛЏФЦКЭЧтбѕЛЏМиЭЈГЃЗжБ№ГЦЮЊПСадФЦ(гжУћЩеМю)КЭПСадМиЁЃЫќУЧЖМЪЧАзЩЋОЇзДЙЬЬхЃЌОпгаНЯЕЭЕФШлЕуЁЃГ§ЧтбѕЛЏяЎЭтЃЌЦфгрМюН№ЪєЕФЧтбѕЛЏЮяЖМвзШмгкЫЎЃЌВЂЗХГіДѓСПЕФШШЁЃдкПеЦјжаШнвзЮќЪЊГБНтЃЌЫљвдЙЬЬхNaOHЪЧГЃгУЕФИЩдяМСЁЃЫќУЧЛЙШнвзгыПеЦјжаЕФCO2ЗДгІЖјЩњГЩЬМЫсбЮЃЌЫљвдвЊУмЗтБЃДцЁЃЕЋдкNaOHБэУцзмФбУтвЊНгДЅПеЦјЖјДјгавЛаЉNa2CO3ЃЌШчЙћдкЛЏбЇЗжЮіЙЄзїжаашвЊВЛКЌNa2CO3ЕФNaOHШмвКЃЌПЩЯШХфжЦNaOHЕФБЅКЭШмвКЃЌNa2CO3вђВЛШмгкБЅКЭЕФNaOHШмвКЖјГСЕэЮіГіЃЌОВжУШЁЩЯВуЧхвКЃЌгУжѓЗаКѓРфШДЕФаТЯЪЫЎЯЁЪЭЕНЫљашЕФХЈЖШМДПЩЁЃМюН№ЪєЧтбѕЛЏЮяЕФЭЛГіЛЏбЇаджЪЪЧЧПМюадЁЃЫќУЧЕФЫЎШмвККЭШлШкЮяЃЌМШФмШмНтФГаЉН№ЪєМАЦфбѕЛЏЮяЃЌвВФмШмНтФГаЉЗЧН№УёМАЦфбѕЛЏЮяЁЃ
2A1+2NaOH+6H2OЃН2Na[Al(OH)4]+3H2Ёќ
A12O3+2NaOHШлШк2NaAlO3+H2O
Si+2NaOH+H2O=Na2SiO3+2H2Ёќ
SiO2+2NaOHЃНNa2SiO3+H2O
вђЮЊЧтбѕЛЏФЦЁЂЧтбѕЛЏМивзгкШлЛЏЃЌгжОпгаШмНтФГаЉН№ЪєбѕЛЏЮяЁЂЗЧН№ЪєбѕЛЏЮяЕФФмСІЃЌвђДЫЙЄвЕЩњВњКЭЗжЮіЙЄзїжаГЃгУгкЗжНтПѓЪЏЁЃШмШкЕФЧтбѕЛЏФЦИЏЪДадИќЧПЃЌЙЄвЕЩЯШлЛЏЧтбѕЛЏФЦвЛАугУж§ЬњШнЦїЃЌдкЪЕбщЪвПЩгУвјЛђФјЕФЦїУѓЁЃЧтбѕЛЏФЦФмИЏЪДВЃСЇЃЌЪЕбщЪвЪЂЧтбѕЛЏФЦШмвКЕФЪдМСЦПЃЌгІгУЯ№ЦЄШћЃЌЖјВЛФмгУВЃСЇШћЃЌЗёдђДцЗХЪБМфНЯГЄЃЌNaOHОЭКЭЦППкВЃСЇжаЕФжївЊГЩЗжSiO2ЗДгІЖјЩњГЩеГадЕФNa2SiO3ЖјАбВЃСЇШћКЭЦППкеГНсдквЛЦ№ЁЃЧтбѕЛЏФЦЪЧвЛжжживЊЕФЛЏЙЄЛљБОдСЯЃЌдкЙЄвЕКЭПЦбЇбаОПЩЯгаКмЖрживЊгУЭОЃЌЫќдкЙЄвЕЩЯЪЧгУЕчНтЪГбЮЫЎШмвКЕФЗНЗЈжЦБИЕФЁЃШчашгУЩйСПЕФЧтбѕЛЏФЦЁЂвВПЩгУПСЛЏЗЈжЦБИЁЃМДгУЯћЪЏЛвЛђЪЏЛвШщгыЬМЫсФЦЕФХЈШмвКЗДгІЃК
Na2CO3+CaЃЈOHЃЉ2ЃНCaCO3Ё§+2NaOH
ЯТУцзХжиЬжТлвЛЯТМюН№ЪєКЭМюЭСН№ЪєЧтбѕЛЏЮяШмНтадКЭМюадЕФБфЛЏЙцТЩЃК
вЛЁЂШмНтЖШЕФБфЛЏ
МюН№ЪєЧтбѕЛЏЮядкЫЎаЁЕФШмНтЖШКмДѓ(LiOHР§Эт)ЃЌВЂШЋВПЕчРыЁЃМюЭСН№ЪєЧтбѕЛЏЮяЕФШмНтЖШБШМюН№ЪєЧтбѕЛЏЮяЕФШмНтЖШаЁЕУЖрЁЃДгБэ17ЁЊ5ПЩвдПДГіЃЌЭЌзхдЊЫиЕФЙ§бѕЛЏЮяЕФШмНтЖШДгЩЯЕНЯТЪЧж№НЅдіДѓЕФЁЃвђЮЊДѓЖрЪ§ЧщПіЯТЃЌРызгЛЏКЯЮяЕФШмНтЖШгыРызгЪЦЃЈZ/rЃЉГЩЗДБШЃЌ(ЯъМћЕкЪЎЮхеТЕкЫФНк)ЁЃМюН№ЪєЧтбѕЛЏЮяДгLiOHЕНCsOHЫцзХбєРызгАыОЖЕФдіДѓЃЌбєРызгКЭвѕРызгжЎМфЕФЮќв§СІж№НЅМѕаЁЃЌROHОЇИёгњРДгњШнвзБЛЫЎЗжзгАбЫќУЧВ№ПЊЁЃЭЌЁЊжмЦкжаЃЌМюЭСН№ЪєРызгБШМюН№ЪєРызгаЁЃЌЖјЧвДјСНИіе§ЕчКЩЃЌвђДЫЫЎЗжзгОЭВЛШнвзНЋЫќУЧВ№ПЊЃЌШмНтЖШОЭаЁЕУЖрЁЃ
ЖўЁЂМюадЕФБфЛЏ
МюН№ЪєКЭМюЭСН№ЪєЧтбѕЛЏЮяМюадГЪЯжгаЙцТЩадЕФБфЛЏЁЃвЛАуЧтбѕЛЏЮяКЭКЌбѕЫсЕФЫсМюадЧПШѕПЩгУІЕ1/2жЕЕФДѓаЁРДХаЖЯЃЈЗчЕкЪЎЮхеТЃЌЕкШ§НкЃЉЁЃЕБН№ЪєРызг(R)ЕФЕчзгЙЙаЭЯрЭЌЪБЃЌдђІЕ1/2жЕгњаЁЃЌМюадгњЧПЃЛДгБэ17ЁЊ6ЫљБэЪОЕФІЕ1/2жЕПЩжЊBe(OH)2ЪЧСНадЧтбѕЛЏЮяЃЌЦфгрМюЭСН№ЪєЧтбѕЛЏЮяОљЮЊМюадЧтбѕЛЏЮяЃЌЖјЧвМюадвРBeЕНBaЕФЫГађЖјдіЧПЁЃ
гЩБэl7ЁЊ6ПЩжЊЃЌЭЌзхдЊЫиЕФЧтбѕЛЏЮяЁЃгЩгкRЕФЕчзгВуЙЙаЭКЭЕчКЩЪ§ОљЯрЭЌЃЌЦфМюадЧПШѕЕФБфЛЏЃЌжївЊШЁОігкРызгАыОЖЕФДѓаЁЁЃЫљвдМюН№ЪєЁЂМюЭСН№ЪєЧтбѕЛЏЮяЕФМюадЃЌОљЫцRРызгАыОЖЕФдіДѓЖјдіЧПЃЌШєАбетСНзхЭЌжмЦкЕФЯрСкСНИідЊЫиЕФЧтбѕЛЏЮяМгвдБШНЯЃЌМюадЕФБфЛЏЙцТЩПЩвдИХРЈШчЯТЃК
ДгЩЯЕНЯТМюаддіЧПЃЛДгзѓЕНгвМюадМѕШѕЁЃ
гЩЩЯЪіПЩжЊЃКетСНзхдЊЫиЧтбѕЛЏЮяМюадЧПШѕЕФБфЛЏЙцТЩКЭдкЫЎжаШмНтЖШЕФБфЛЏЙцТЩЪЧвЛжТЕФЁЃЮЊЪВУДМюН№ЪєЧтбѕЛЏЮяЕФМюадЬиБ№ЧПЃПетвЛЗНУцгЩгкЫќУЧдкЫЎШмвКжагаНЯДѓЕФШмНтЖШЃЌПЩвдЕУЕНХЈЖШНЯДѓЕФШмвКЃЛСэвЛЗНУцЃЌЫќУЧдкЫЎШмвКжаМИКѕШЋВПЕчРыЃЌвђДЫПЩвдЕУЕНИпХЈЖШЕФOH-РызгЃЌOH-РызгХЈЖШгњДѓЃЌМюадОЭгњЧПЁЃвђДЫМюН№ЪєЕФЧтбѕЛЏЮяЪЧзюЧПЕФМюЁЃМюЭСН№ЪєЕФЧтбѕЛЏЮяШмНтЖШБШМюН№ЪєаЁЕУЖрЃЌМюЪНЕчРыГЬЖШБШНЯВюЃЌЫљвдЦфМюадБШМюН№ЪєЧтбѕЛЏЮяРДЫЕвЊШѕвЛаЉЁЃ
МюН№ЪєКЭМюЭСН№ЪєЕФживЊЕФбЮРрМАЦфаджЪЃКМюН№ЪєКЭМюЭСН№ЪєЕФГЃМћбЮРргаТБЛЏЮяЁЂЬМЫсбЮЁЂЯѕЫсбЮЁЂСђЫсбЮКЭСђЛЏЮяЕШЃЌЯТУцЬжТлЫќУЧЕФЙВадКЭвЛаЉЬиадЃЌВЂМђЕЅНщЩмМИжжживЊЕФбЮЁЃ
МюН№ЪєбЮРрЕФзюДѓЬиеїЪЧвзШмгкЫЎЃЌВЂЧвдкЫЎжаЭъШЋЕчРыЃЌЫљгаМюН№ЪєРызгЖМЪЧЮоЩЋЕФЁЃжЛгаЩйЪ§МюН№ЪєбЮЪЧФбШмЕФЁЃЫќУЧЕФФбШмбЮвЛАуЖМЪЧгЩДѓЕФвѕРызгзщГЩЃЌЖјЧвМюН№ЪєРызгдНДѓЁЂФбШмбЮЕФЪ§ФПвВдНЖрЁЃ
ФбШмФЦбЮгаАзЩЋСЃзДЕФСљєЧЛљЬрЫсФЦNa[Sb(OH)6]ЃЌДзЫсЫЋбѕгЫѕЃаПФЦNaAcЁЄZn(Ac)2ЁЄ3UO2(Ac)2ЃЌ9H2OЮЊЛЦТЬЩЋНсОЇЁЃФбШмЕФМибЮЩдЖрЃЌгаЃКИпЪдЫсМиKClO4(АзЩЋ)ЃЌЫФБНХ№ЫсМиKB(C6H5)4](АзЩЋ)ЃЌШїЪЏЫсЧтМиKHC4H4O6(АзЩЋ)ЃЌСљТШВЌЫсМиK2[PtCl4](ЕЛЦЩЋ)ЃЌюмбЧЯѕЫсФЦМиK2Na[Co(NO2)6](ССЛЦЩЋ)ЃЌФЦЁЂМиЕФвЛаЉФбШмбЮГЃгУдкМјЖЈФЦЁЂМиРызгЁЃ
МюЭСН№ЪєбЮРрЕФживЊЬиеїЪЧЫќУЧЕФЮЂШмадЁЃГ§ТШЛЏЮяЁЂЯѕЫсбЮЁЂСђЫсУОЁЂИѕЫсУОвзШмгкЫЎЭтЃЌЦфгрЕФЬМЫсбЮЁЂСђЫсбЮЁЂВнЫсбЮЁЂИѕЫсбЮЕШНдФбШмЁЃСђЫсбЮКЭИѕЫсбЮЕФШмНтЖШCaЁЂSrЁЂBaЕФЫГађНЕЕЭЁЃИяЫсИЦЕФШмНтЖШЪЧЫљгаИЦбЮжазюаЁЕФЃЌвђДЫдкжиСПЗжЮіжаПЩгУЫќРДВтЖЈИЦЁЃ
МюН№ЪєКЭМюЭСН№ЪєЬМЫсбЮШмНтЖШЕФВюБ№вВГЃгУРДЗжРыNa+ЁЂK+КЭCa2+ЁЂBa2+ЁЃ
ФЦбЮКЭМибЮаджЪКмЯрЫЦЃЌЕЋвВгаВюБ№ЃЌживЊЕФгаШ§ЕуЃК
1)ЃЎШмНтЖШФЦЁЂМибЮЕФШмНтЖШЖМБШНЯДѓЃЌЯрЖдЫЕРДЃЌФЦбЮИќДѓаЉЁЃНіNaHCO3ЕФШмНтЖШВЛДѓЃЌNaClЕФШмНтЖШЫцЮТЖШЕФБфЛЏВЛДѓЃЌетЪЧГЃМћЕФФЦбЮжаШмНтадНЯЬиЪтЕФЁЃ
2).ЮќЪЊадФЦбЮЕФЮќЪЊадБШЯргІЕФМибЮЧПЁЃвђДЫЃЌЛЏбЇЗжЮіЙЄзїжаГЃгУЕФБъзМЪдМСаэЖрЪЧМибЮЃЌШчгУСкБНЖўМзЫсЧтМиБъЖЈМювКЕФХЈЖШЃЌгУжиИѕЫсМиБъЖЈЛЙдМСШмвКЕФХЈЖШЁЃдкХфжЦеЈвЉЪБгУKNO3ЛђKClO3ЃЌЖјВЛгУЯргІЕФФЦбЮЁЃ
3)ЃЎНсОЇЫЎКЌНсОЇЫЎЕФФЦбЮБШМибЮЖрЁЃШчNa2SO4ЁЄ10H2OЁЂK2SO4ЁЂNa2HPO4ЁЄ10H2OЕШЁЃ
ОјДѓЖрЪ§МюН№ЪєКЭМюЭСН№ЪєЕФбЮЪЧРызгаЭОЇЬхЃЌОЇЬхДѓЖрЪ§ЪєNaClаЭЃЌяЄЕФТБЛЏЮяЪЧCsClаЭНсЙЙЁЃЫќУЧЕФШлЕуОљНЯИпЁЃгЩгкLi+ЁЂBe2+РызгзюаЁЃЌМЋЛЏзїгУНЯЧПЃЌВХЪЙЕУЫќУЧЕФЦфаЉбЮ(ШчТБЛЏЮя)ОпгаНЯУїЯдЕФЙВМладЁЃMg2+бЮвВгавЛаЉЪЧЙВМладЕФЁЃ
вЛАуРДЫЕЃЌРызггњаЁЃЌЫќЫљДјЕФЕчКЩгњЖрЃЌдђзїгУгкЫЎЗжзгЕФЕчГЁгњЧПЃЌЫќЕФЫЎКЯШШгњДѓЁЃМюН№ЪєРызгЪЧзюДѓЕФе§РызгЃЌРызгЕчКЩзюЩйЃЌдЫќЕФЫЎКЯШШГЃаЁгкЦфЫќРызгЁЃ
МюН№ЪєРызгЕФЫЎКЯФмСІДгLiЁњCsЪЧНЕЕЭЕФЃЎетвВЗДгГдкбЮРраЮГЩНсОЇЫЎКЯЮяЕФЧуЯђЩЯЁЃМИКѕЫљгаЕФяЎбЮЪЧЫЎКЯЕФЃЌФЦбЮдМга75%ЪЧЫЎКЯЕФЃЌМибЮга25%ЪЧЫЎКЯЮяЃЌяЈбЮКЭяЄбЮНігаЩйЪ§ЪЧЫЎКЯбЮЁЃдкГЃМћЕФМюН№ЪєбЮжаЃЌТБЛЏЮяДѓЖрЪЧЮоЫЎЕФЃЌЯѕЫсбЮжажЛгаяЎаЮГЩЫЎКЯЮяЃЌLiNO3ЁЄH2OКЭLiNO3ЁЄ3H2OЃЌСђЫсбЮжЛгаLi2SO4ЁЄH2OКЭNa2SO4ЁЄ10H2OЃЌЬМЫсбЮжаГ§Li2CO3ЮоЫЎКЯЮяЭтЃЌЦфгрНдгаВЛЭЌаЮЪНЕФЫЎКЯЮяЃЌЦфЫЎЗжзгЪ§ЗжБ№ЮЊЃК
Na2CO3K2CO3Rb2CO3Cs2CO3
1ЁЂ7ЁЂ101ЁЂ51ЁЂ53ЁЂ5
аЮГЩИДбЮЕФФмСІЃКГ§яЎвдЭтЃЌМюН№ЪєЛЙФмаЮГЩвЛЯЕСаИДбЮЁЃИДбЮгавдЯТМИжжРраЭЃК
ЙтТБЪЏРрЃЌЭЈЪНЮЊMЂёC1•��������MgCl2•��������H2OЃЌЦфжаMЂёЃНK+ЁЂRb+ЁЂCs+ЃЌШчЙтТБЪЏKCl•��������MgCl2ЁЄ6H2OЃЛ
ЭЈЪНЮЊMЂё2SO4•��������MgSO4•��������6H2OЕФЗЏРрЃЌЦфжаMЂёЃНK+ЁЂRb+ЁЂCs+ЃЌШчШэМиУОЙЎK2SO4ЁЄMgSO4ЁЄ6H2OЃЛ
ЭЈЪНЮЊMЂёMЂѓ(SO4)2ЁЄ12H2OЕФЗЏРрЃЌЦфжаMЂё=Na+ЁЂK+ЁЂRb+ЁЂCs+ЃЌMЂѓЃНA13+ЁЂCr3+ЁЂFe3+ЁЂCo3+ЁЂGa3+ЁЂV3+ЕШРызгЃЌШчУїЗЏKAl(SO4)2ЁЄ12H2OЁЃ
ШШЮШЖЈадЃКвЛАуМюН№ЪєбЮОпгаНЯИпЕФШШЮШЖЈадЁЃТБЛЏЮядкИпЮТЪБЛгЗЂЖјФбЗжНтЁЃСђЫсбЮдкИпЮТЯТМШФбЛгЗЂЃЌгжФбЗжНтЁЃЬМЫсбЮГ§Li2CO3дк1543KвдЩЯЗжНтЮЊLi2OКЭCO2ЭтЃЌЦфгрИќФбЗжНтЁЃЮЈгаЯѕЫсбЮШШЮШЖЈадНЯЕЭЃЌМгШШЕНвЛЖЈЮТЖШОЭПЩЗжНтЃЛ
МюЭСН№ЪєЕФТБЛЏЮяЁЂСђЫсбЮЁЂЬМЫсбЮЖдШШвВНЯЮШЖЈЃЌЕЋЫќУЧЕФЬМЫсбЮШШЮШЖЈадНЯМюН№ЪєЬМЫсбЮвЊЕЭЁЃ
МюЭСН№ЪєЬМЫсбЮШШЮШЖЈадЕФЙцТЩвВПЩгУРызгМЋЛЏЕФЙлЕуРДЫЕУїЁЃ
МИжжживЊЕФбЮЃКТБЛЏюыЪЧЙВМлаЭОлКЯЮяЃЈBeX2ЃЉn,ВЛЕМЕчЁЂФмЩ§ЛЊЃЌеєЦјжагаBeCl2КЭ(BeCl2)2ЗжзгЁЃТБЛЏЮяжагУЭОзюЙуЕФЪЧТШЛЏФЦЃЌгаКЃбЮЁЂбвбЮКЭОЎбЮЕШЁЃТШЛЏФЦГ§ЙЉЪГгУЭтЃЌЫќЪЧжЦШЁН№ЪєФЦЁЂЧтбѕЛЏФЦЁЂЬМЫсФЦЁЂТШЦјКЭбЮЫсЕШЖржжЛЏЙЄВњЦЗЕФЛљБОдСЯЁЃБљбЮЛьКЯЮяПЩзїЮЊжТРфМСЁЃЮоЫЎТШЛЏУОЪЧжЦШЁН№ЪєУОЕФдСЯЃЌЙтТБЪЏКЭКЃЫЎЪЧШЁЕУТШЛЏУОЕФжївЊзЪдДЁЃТШЛЏУОГЃПіЯТвдMgCl2ЁЄ6H2OаЮЪНДцдкЃЌгУМгШШЫЎКЯЮяЕФЗНЗЈВЛФмЕУЕНЮоЫЎбЮЃЌвђЮЊЫќЛсЫЎНтЃК
MgCl2ЁЄ6H2O>408KMg(OH)Cl+HCl+5H2O
Mg(OH)C770KMgO+HCl
вЊЕУЕНЮоЫЎЕФТШЛЏУОЃЌБиаыНЋСљВЛТШЛЏУОдкИЩдяЕФТШЛЏЧтЦјСїжаМгШШЭбЫЎЁЃЙЄвЕЩЯГЃгУдкИпЮТЯТЭЈТШЦјгкНЙЬПКЭбѕЛЏУОЕФЛьКЯЮяжЦШЁЁЃТШЛЏУОгаЮќГБадЃЌЦеЭЈЪГбЮЕФГБНтОЭЪЧКЌгаТШЛЏУОжЎЙЪЁЃСљЫЎТШЛЏИЦШШжС473KЪЇШЅЫЎЖјГЩЖўЫЎТШЛЏИЦЃЌЮТЖШИпгк533KЭъШЋЭбЫЎаЮГЩАзЩЋЖрПзЕФТШЛЏИЦЃЌДЫетГЬгаЩйаэЫЎНтЗДгІЗЂЩњЃЌЙЪЮоЫЎТШЛЏИЦжаГЃКЌгаЮЂСПбѕЛЏИЦЁЃЮоЫЎТШЛЏИЦгаКмЧПЕФЮќЫЎадЃЌЪЧвЛжжживЊЕФИЩдяМСЁЃгЩгкЫќФмгыЦјЬЌАБКЭввДМаЮГЩМгГЩЮяЃЌЫљвдВЛФмгУгкИЩдяАБЦјКЭввДМЁЃТШЛЏИЦКЭБљЃЈ1.44ЃК1ЃЉЕФЛьКЯЮяЪЧЪЕбщЪвГЃгУЕФжТРфМСЃЌПЩЛёЕУ218KЕФЕЭЮТЁЃТШЛЏБЕЮЊЮоЩЋЕЅаБОЇЬхЃЌвЛАуЮЊЫЎКЯЮяЖўЫЎТШЛЏБЕЁЃМгШШжС400KБфЮЊЮоЫЎбЮЁЃТШЛЏБЕгУгквНвЉЁЂУ№ЪѓМСКЭМјЖЈСђЫсИљРызгЕФЪдМСЁЃТШЛЏБЕПЩШмгкЫЎЁЃПЩШмадБЕбЮЖдШЫЁЂаѓЖМгаКІЃЌЖдШЫжТЫРСПЮЊ0.8gЃЌЧаМЩШыПкЁЃ
ЗњЛЏИЦЃЈгЉЪЏЃЉЪЧжЦШЁHFКЭF2ЕФживЊдСЯЁЃдквБН№ЙЄвЕжагУзїжњШлМСвВгУгкжЦзїЙтбЇВЃСЇКЭЬеДЩЕШЁЃ
ГЃгУЕФгЋЙтЕЦжаЭПгагЋЙтВФСЯ3Ca3(PO4)2Ca(F,Cl)2КЭЩйСПSb3+ЁЂMn2+ЕФЛЏКЯЮяЃЌТБСзЫсИЦГЦЮЊФИЬхЃЌSb3+ЁЂMn2+РызгЮЊМЄЛюМСЃЌгУзЯЭтЙтМЄЗЂКѓЃЌЗЂГігЋЙтЁЃ
МюН№ЪєЬМЫсбЮгаСНРрЃКе§бЮКЭЫсЪНбЮЁЃЬМЫсФЦЫзГЦЫеДђЛђДПМюЃЌЦфЫЎШмвКвђЫЎНтЖјГЪМюадЁЃЫќЪЧвЛжжживЊЕФЛЏЙЄдСЯЁЃЬМЫсЧтФЦЫзГЦаЁЫеДђЃЌЦфЫЎШмвКГЪШѕМюадЃЌжївЊгУгквНвЉКЭЪГЦЗЙЄвЕЃЌьбЩеЬМЫсЧтФЦПЩЕУЕНЬМЫсФЦЁЃ
СљЫЎЬМЫсИЦЮЊЮоЩЋЕЅаБОЇЬхЃЌФбШмгкЫЎЃЌвзШмгкЫсКЭТШЛЏяЇШмвКЃЌгУгкжЦЖўбѕЛЏЬМНЭЗлКЭЭПСЯЕШЁЃЬМЫсИЦЮЊЮоЩЋаБЗНОЇЬхЃЌШШжС1000KзЊБфЮЊЗННтЪЏЁЃ
ЯѕЫсМидкПеЦјжаВЛЮќГБЃЌдкМгШШЪБгаЧПбѕЛЏадЃЌгУРДжЦКкЛ№вЉЁЃЯѕЫсМиЛЙЪЧКЌЕЊЗЪЁЂМиЕФгХжЪЛЏЗЪЁЃ
Na2SO4ЁЄ10H2OЫзГЦУЂЯѕЃЌгЩгкЫќгаКмДѓЕФШлЛЏШШЃЌЪЧвЛжжНЯКУЕФЯрБфжќШШВФСЯЕФжївЊзщЗжЃЌПЩгУгкЕЭЮТжќДцЬЋбєФмЁЃАзЬьЫќЮќЪеЬЋбєФмЖјШлШкЃЌвЙМфРфШДНсОЇОЭЪЭЗХГіШШФмЁЃЮоЫЎСђЫсФЦЫзГЦдЊУїЗлЃЌДѓСПгУгкВЃСЇЁЂдьжНЁЂЫЎВЃСЇЁЂЬеДЩЕШЙЄвЕжаЃЌвВгУгкжЦСђЛЏФЦКЭСђДњСђЫсФЦЕШЁЃ
CaSO4ЁЄ2H2OЫзГЦЩњЪЏИрЃЌМгШШжС393KзѓгвЫќВПЗжЭбЫЎЖјГЩЪьЪЏИрCaSO4ЁЄ1/2H2OЃЌетИіЗДгІЪЧПЩФцЕФЃК
2CaSO4ЁЄ2H2O393K2CaSO4ЁЄ1/2H2O+3H2O
ЪьЪЏИргыЫЎЛьКЯГЩК§зДКѓЗХжУвЛЖЮЪБМфЛсБфГЩЖўЫЎКЯбЮЃЌетЪБж№НЅгВЛЏВЂХђеЭЃЌЙЪгУвджЦФЃаЭЁЂЫмЯёЁЂЗлБЪКЭЪЏИрБСДјЕШЁЃЪЏИрЛЙЪЧЩњВњЫЎФрЕФдСЯжЎвЛКЭЧсжЪНЈжўВФСЯЁЃАбЪЏгЂжгИрМгШШЕН773KвдЩЯЃЌЕУЕНЮоЫЎЪЏИрЃЌЫќВЛФмгыЫЎЛЏКЯЁЃ
жиОЇЪЏСђЫсБЕЪЧжЦБИЦфЫќБЕРрЛЏКЯЮяЕФдСЯЁЃНЋжиОЇЪЏЗлгыУКЗлЛьКЯЃЌдкИпЮТЯТЃЈ1173K-1473KЃЉьбЩеЛЙдГЩПЩШмадСђЛЏБЕЁЃ
бЮЫсгыСђБЕЗДгІЃЌжЦЕУТШЛЏБЕЁЃЭљСђЛЏБЕШмвКжаЭЈШыЖўбѕЛЏЬМЃЌдђЕУЬМЫсБЕЁЃ
жиОЇЪЏПЩзїАзЩЋЭПСЯЃЈБЕАзЃЉЃЌдкЯ№НКЁЂдьжНЙЄвЕжазїАзЩЋЬюСЯЁЃСђЫсБЕЪЧЮЈвЛЮоЖОБЕбЮЃЌгУгкГІЮИЯЕЭГXЩфЯпдьгАМСЁЃ
ЦпЫЎСђЫсУОЮЊЮоЩЋаБЗНОЇЬхЁЃШШжС350KЪЇШЅСљЗжзгЫЎЃЌдк520KБфЮЊЮоЫЎбЮЁЃСђЫсУОЮЂШмгкДМЃЌВЛШмгкввЫсКЭБћЭЊЃЌгУзїУНШОМСЁЂаКбЮЃЌвВгУгкдьжНЁЂЗФжЏЁЂЗЪдэЁЂЬеДЩЁЂгЭЦсЙЄвЕЁЃ
Н№ЪєРызгжаЃЌФЦЃЌМиЃЌяЈЃЌяЄРызгвђЕчКЩЩйЃЌАыОЖДѓКЭУЛгаОЇЬхГЁЮШЖЈЛЏаЇгІЃЌаЮГЩвЛАуХфКЯЮяЕФЧуЯђзюаЁЁЃЫќУЧФмгыХфЮЛФмСІНЯЧПЕФђќКЯМСзїгУЃЌЩњГЩђќКЯЮяЁЃЫљгаМюН№ЪєЖМПЩвдКЭЫЎбюШЉзїгУЃЌЩњГЩХфЮЛЪ§ЮЊ4ЕФХфЮЛЛЏКЯЮяЁЃ
МюЭСН№ЪєРыжаюыРыАыОЖзюаЁЃЌЪЧНЯЧПЕФЕчзгЖдНгЪмЬхЃЌФмаЮГЩНЯЖрЕФХфКЯЮяЁЃ
ИЦЁЂяШЁЂБЕЕФРызгАыОЖНЯДѓЃЌЩњГЩХфКЯЮяЕФФмСІНЯШѕЃЌБЕРызгЕФХфКЯЮяКмЩйЁЃ
яЎЁЂюыЕФЬиЪтаджЪ-ЖдНЧЯпЙцдђЃКдкжмЦкБэжаЃЌгаМИЖдДІгкЯрСкСНзхЕФЖдНЧЯпЩЯЕФдЊЫиЃЌМДLiЁЊMgЃЌBeЁЊA1ЃЌBЁЊSiЃЌЫќУЧОпгааэЖрЯрЫЦЕФаджЪЃЌШЫУЧГЦжЎЮЊаБЯпЙиЯЕЛђЖдНЧЯпЙиЯЕЁЃ
яЎгыУОЕФЯрЫЦадЃК
ЂХLi+(66pm)ЁЂMg2+(65pm)АыОЖЯрНќ
(2)ЕЅжЪгыбѕзїгУЩњГЩе§ГЃбѕЛЏЮя
ЂЦЕЅжЪгыЫЎЗДгІОљНЯЛКТ§ЃЌЧтбѕЛЏЮяОљЮЊжаЧПМюЃЌЧвЫЎжаШмНтЖШВЛДѓ
ЂЧЗњЛЏЮяЁЂЬМЫсбЮЁЂСзЫсбЮОљФбШм
ЂШТШЛЏЮяОљФмШмгкгаЛњШмМСжа
ЂЩЬМЫсбЮЪмШШЗжНтЃЌВњЮяЮЊЯргІбѕЛЏЮя
2.юыгыТСЕФЯрЫЦадЃК
ЂХЖМЪЧСНадН№ЪєЃЌЦфбѕЛЏЮяЁЂЧтбѕЛЏЮяОљЮЊСНадЃЌбѕЛЏЮяШлЕуИпЁЂгВЖШДѓЃЌЧтбѕЛЏЮяФбШмгкЫЎЃЌBe2+ЁЂAl3+бЮвзЫЎНт
(2)БъзМЕчМЋЕчЪЦЯрНќЃЌЃХo(Be2+/Be)=-1.97v,ЁЁЃХo(Al3+/Al)=-1.68v
(3)BeCl2ЁЂAlCl3ЮЊЙВМлЛЏКЯЮяЃЌвзОлКЯЃЌПЩШмгкгаЛњШмМСЃЛBe2+ЁЂAl3+гыFЃЖМПЩЩњГЩЗњЕФХфКЯЮяЁЊЁЊBeF4-ЃЌAlF63-
ЂШЕЅжЪОљФмБЛРфЕФХЈHNO3ЖлЛЏ
ЕкЪЎвЛеТ ТБЫиКЭбѕзхдЊЫи
pЧјдЊЫиИХЪі
1. ИїзхдЊЫиаджЪгЩЩЯЕНЯТЕФБфЛЏЙцТЩГіЯжЭЛБф
2. ЖржжбѕЛЏЬЌ(вђЮЊМлЕчзгЙЙаЭЮЊns2np1-5)
3. ЕчИКадДѓЃЌаЮГЩЙВМлЛЏКЯЮя
ЭЛБфЃК
1. ЕквЛХХдЊЫиЗДГЃадЃК(жЛга2s,2pЙьЕР) аЮГЩХфКЯЮяЪБЃЌХфЮЛЪ§зюЖрВЛГЌЙ§4
ЕквЛХХдЊЫиЕЅМќМќФмаЁгкЕкЖўХХдЊЫиЕЅМќМќФм
E(N-N)=159 kJЁСmol-1 E(O-O)=142 kJЁСmol-1 E(F-F)=158 kJЁСmol-1
E(P-P)=209 kJЁСmol-1 E(S-S)=264 kJЁСmol-1 E(Cl-Cl)=244 kJЁСmol-1
2. жаМфХХдЊЫивьбљад(dЧјВхШы) Р§ШчЃКфхЫсЁЂИпфхЫсбѕЛЏадОљБШЦфЫќТБЫсЁЂИпТБЫсЧПЁЃ
3. зюКѓШ§ИідЊЫиаджЪЛКТ§ЕиЕнБф(ячЯЕЪеЫѕ)ЁЃ
ЖржжбѕЛЏЬЌЃК
вђЮЊМлЕчзгЙЙаЭЮЊЃКns2np1-5ЃЌpЧјдЊЫигаЖрИіМлЕчзгЃЌЫљвдПЩвдаЮГЩЖржжбѕЛЏЬЌЁЃ
Р§ШчЃКТШга+1ЃЌ+3ЃЌ+5ЃЌ+7ЃЌ-1ЃЌ0ЕШбѕЛЏЬЌЁЃ
ЖдгкЭЌзхдЊЫиЖјбдДгЩЯЕНЯТЃЌЫцзХдзгађЪ§ЕФдіМгЕЭбѕЛЏЬЌЛЏКЯЮяБШИпбѕЛЏЬЌЛЏКЯЮяЮШЖЈЃЌетжжЯжЯѓГЦЮЊЖшадЕчзгЖдаЇгІЁЃ
Р§ЃКSn(II)<Sn(IV) Pb(II)>Pb(IV)
Ёь11.2 ТБЫи
ТБЫиЪЧдЊЫижмЦкЯЕЕкЂїAзхдЊЫиЃЌАќРЈЗњЁЂТШЁЂфхЁЂЕтЁЂэСЁЃ
ТБзхдЊЫиЕФаджЪБфЛЏШчЯТЃК
ТБЫидЊЫиЗћКХ F Cl Br I
МлЕчзгЙЙаЭ 2s22p5 3s23p5 4s24p5 5s25p5
ЙЋМлАыОЖ/pm 66 99 114 133
ЕчИКад 3.98 3.16 2.96 2.66
ЕчРыФм/ЃЈkJЁЄmol-1ЃЉ 1687 1257 1146 1015
11.2.2 ТБЫиЕЅжЪ
ЂБТБЫиЕЅжЪЕФЮяРэаджЪ

ЂВТБЫиЕЅжЪЕФЛЏбЇаджЪ
ТБЫиЪЧКмЛюЦУЕФЗЧН№ЪєдЊЫиЁЃТБЫиЕЅжЪОпгаКмЧПЕФбѕЛЏадЃЌФмгыДѓЖрЪ§дЊЫижБНгЛЏКЯЁЃ
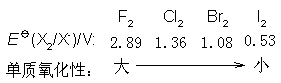
Р§ШчЃКЮЛгкЧАУцЕФТБЫиЕЅжЪПЩвдбѕЛЏКѓУцТБЫиЕФвѕРызгЁЃ
Cl2 + 2Br- Ёњ 2Cl- + Br2
Ёя ТБЫигыЫЎЗДгІЗжЮЊСНРрЃК
бѕЛЏЗДгІЃК 2X2 + 2H2O Ёњ 4X- + 4H+ + O2
МЄСвГЬЖШЃКF2ЃОCl2ЃОBr2ЃЌЕтВЛЗЂЩњДЫРрЗДгІЁЃ
ЦчЛЏЗДгІЃК ![]()
![]()
![]()
![]()
ПЩМћЃЌЗДгІНјааЕФГЬЖШCl2ЃОBr2ЃОI2,ЗњжЛЗЂЩњЕквЛРрЗДгІЁЃЭЈГЃЫљгУЕФТШЫЎЁЂфхЫЎЁЂЕтЫЎжївЊГЩЗжЪЧЕЅжЪЁЃ
Ёя ТБЫидкМюадЬѕМўЯТЗЂЩњСНРрЦчЛЏЗДгІЃК X2 + 2OHЃ Ёњ XЃ + XOЃ + H2O
3X2 + 6OHЃ Ёњ 5XЃ + XO3Ѓ + 3H2O
ВЛЭЌдЊЫиЕЅжЪЗЂЩњЦчЛЏЗДгІЕФЬѕМўМАжївЊВњЮяМћЯТБэЃК
ГЃЮТ МгШШ ЕЭЮТ
Cl2 ClO- ClO3- ClO- pHЃО4
Br2 BrO3- BrO3- BrO-(0Ёц) pHЃО6
I2 IO3- IO3- IO3 pHЃО9
11.2.3 ТБЫиЕФЧтЛЏЮя
ЂБТБЫиЕФЧтЛЏЮяИХЪі
ТБЫиЕФЧтЛЏЮяГЦЮЊТБЛЏЧтЃЌМДЗњЛЏЧтHFЁЂТШЛЏЧтHClЁЂфхЛЏЧтHBrЁЂЕтЛЏЧтHIЕШЁЃГЃЮТЯТТБЛЏЧтЖМЪЧЮоЩЋЁЂгаДЬМЄадЦјЮЖЕФЦјЬхЁЃТБЛЏЧтвзШмгкЫЎЃЌЦфЫЎШмвКНаЧтТБЫсЁЃГ§ЗњЛЏЧтЭтЃЌЦфЫќЧтЛЏЮяОљЮЊЧПЫсЁЃ
ЂВТШЛЏЧтКЭбЮЫс
жЦБИЃК жБНгКЯГЩЗЈЃК ![]()
ИДЗжНтЗДгІЃК
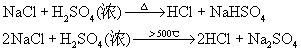
ДПбЮЫсЮЊЮоЩЋШмвКЃЌгаТШЛЏЧтЕФДЬМЄадЦјЮЖЁЃвЛАуХЈбЮЫсЕФХЈЖШдМЮЊ39%ЃЌЯрЕБгк12molЁЄl-1ЃЌУмЖШЮЊ1.19gЁЄcm-3ЁЃЙЄвЕгУЕФбЮЫсЮЊ30%зѓгвЃЌгЩгкДјгадгжЪЖјДјЛЦЩЋЁЃ
гУЭОЃКбЮЫсЪЧживЊЕФЛЏЙЄЩњВњдСЯЃЌГЃгУРДжЦБИН№ЪєТШЛЏЮяЁЂБНАЗКЭШОСЯЕШВњЦЗЁЃбЮЫсдквБН№ЙЄвЕЁЂЪЏгЭЙЄвЕЁЂгЁШОЙЄвЕЁЂЦЄИяЙЄвЕЁЂЪГЦЗЙЄвЕМАдўИжЁЂКИНгЁЂЕчЖЦЁЂЬТДЩЁЂвЉЮяЕШВПУХвВгаЙуЗКЕФгІгУЁЃ
ЂГЗњЁЂфхЁЂЕтЕФЧтЛЏЮя
ЗњЛЏЧтЕФжЦБИ
вЛАугУИДЗжНтЗДгІЗЈЃК
CaF2 + H2SO4(ХЈ) Ёњ CaSO4 + 2HF
ЗњЛЏЧтЪЧЮоЩЋЁЂгаДЬМЄадЦјЮЖВЂОпгаЧПСвИЏЪДадЕФгаЖОЦјЬхЃЌЕБЦЄЗєНгДЅHFЪБЛсв§Ц№ВЛвзШЌгњЕФзЦЩЫЃЛЧтЗњЫсЕФеєЦћЖдЦЄЗєвВгаЭЌбљЕФЮЃКІЃЌвђДЫЃЌЪЙгУЧтЗњЫсЪБгІЬиБ№зЂвтАВШЋЁЃЗњЛЏЧтКЭЧтЗњЫсЖМФмКЭЖўбѕЛЏЙшзїгУЃЌЩњГЩЛгЗЂадЕФЫФЗњЛЏЙшЃК
SiO2 + 4HF Ёњ SiF4 + 2H2O
ЖўбѕЛЏЙшЪЧВЃСЇЕФжївЊГЩЗжЃЌЧтЗњЫсФмИЏЪДВЃСЇЁЃвђДЫЃЌГЃгУЫмСЯШнЦїРДжќДцЧтЗњЫсЁЃ
ЧтЗњЫсПЩвдгУРДПЬЪДВЃСЇЛђШмНтИїжжЙшЫсбЮЁЃ
фхЛЏЧтКЭЕтЛЏЧтЕФжЦБИ
вЛАугУТБЛЏЮяЫЎНтЗЈЃК PBr3 + 3H2O Ёњ H3PO3 + 3HBr
PI3 + 3H3O Ёњ H3PO3 + 3HI
вВПЩжБНггУЫЎКЭТБЫигыСзЛьКЯЮяЗДгІжЦБИТБЛЏЧтЁЃ
2P + 3Br2 + 6H2O Ёњ 2H3PO3 + 6HBr
2P + 3I2 + 6H2O Ёњ 2H3PO3 + 6HI
фхЛЏЧтКЭЕтЛЏЧтЕФжЦБИВЛФмгУЕЅжЪжБНгКЯГЩЗЈЃЌвђфхКЭЕтгыЧтЗДгІЫйЖШЛКТ§ЁЂВњТЪЕЭЖјВЛЪЪКЯЁЃфхЛЏЧтКЭЕтЛЏЧтЕФжЦБИвВВЛФмгУТБЛЏЮягыХЈСђЫсЕФИДЗжНтЗДгІРДжЦБИЃЌвђЮЊHBrЁЂHIгаЯджјЕФЛЙдадЃЌЫќУЧНЋгыХЈСђЫсНјвЛВНЗЂЩњбѕЛЏЛЙдЗДгІЃК
2HBr + H2SO4(ХЈ) Ёњ Br2 + SO2 + 2H2O
8HI + H2SO4(ХЈ) Ёњ 4I2 + H2S + 4H2O
ЫљвдЃЌЪЕМЪЩЯЕУВЛЕНДПЕФфхЛЏЧтКЭЕтЛЏЧтЁЃШчЙћИФгУЮобѕЛЏадЕФИпЗаЕуЫсХЈСзЫсДњЬцХЈСђЫсЃЌПЩвджЦЕУфхЛЏЧтКЭЕтЛЏЧтЁЃ
ЂД ТБЛЏЧтаджЪЕФБШНЯ

11.2.4 ТБЛЏЮяКЭЖрТБЛЏЮя
ЂБТБЛЏЮя
ТБЫиКЭЕчИКадБШЫќаЁЕФдЊЫиЩњГЩЕФЛЏКЯЮяНаТБЛЏЮяЁЃТБЛЏЮяПЩвдЗжЮЊН№ЪєТБЛЏЮяКЭЗЧН№ЪєТБЛЏЮяЃЌИљОнТБЛЏЮяЕФМќаЭЃЌгжПЩвдЗжЮЊРызгаЭТБЛЏЮяКЭЙВМлаЭТБЛЏЮяЁЃ
ЂХН№ЪєТБЛЏЮя
ЫљгаН№ЪєЖМФмаЮГЩТБЛЏЮяЁЃМюН№ЪєЁЂМюЭСН№ЪєвдМАячЯЕЁЂяЙЯЕдЊЫиЕФТБЛЏЮяДѓЖрЪ§ЪєгкРызгаЭЛђНгНќРызгаЭЃЌР§ШчЃКNaXЃЌBaCl2ЃЌLaCl3ЕШЁЃЕБвѕбєРызгМЋЛЏзїгУБШНЯУїЯдЪБЃЌБэЯжГівЛЖЈЕФЙВМладЃЌШчЃКAgClЕШЁЃгааЉИпбѕЛЏжЕЕФН№ЪєТБЛЏЮядђЮЊЙВМлаЭТБЛЏЮяЃЌШчЃЌAlCl3,SnCl4,FeCl3,TiCl4ЕШЁЃ
ВЛЭЌРраЭТБЛЏЮяЃЌаджЪЩЯДцдкВювьЃЌМћЯТБэЃК
ТБЛЏЮяРраЭ РызгаЭ ЙВМлаЭ
ШлЕу Ип ЕЭ
ШмНтад ДѓЖрвзШмгкЫЎ взШмгкгаЛњШмМС
ЕМЕчад ЫЎШмвКЁЂШлШкЕМЕч ЮоЕМЕчад
дкН№ЪєТБЛЏЮяжа,ЖдгІЧтбѕЛЏЮяВЛЪЧЧПМюЕФЖМвзЫЎНтЃЌВњЮяЮЊЧтбѕЛЏЮяЛђМюЪНбЮЁЃашЬиЪтМЧвфЕФгаЃК
SnCl2 + H2O Ёњ Sn(OH)Cl + HCl
SbCl3 + H2O Ёњ SnOCl + 2HCl
BiCl3 + H2O Ёњ BiOCl + 2HCl
ЂЦЗЧН№ЪєТБЛЏЮя
ЗЧН№ЪєХ№ЁЂЬМЁЂЙшЁЂЕЊЁЂСзЕШЖМФмгыТБЫиаЮГЩИїжжЯргІЕФТБЛЏЮяЁЃетаЉТБЛЏЮяЖМЪЧЙВМлаЭЕФЁЃ
ЗЧН№ЪєТБЛЏЮяЫЎНтВњЮявЛАуЮЊСНжжЫсЃЌР§ШчЃКBX3ЃЌSiX4ЃЌPCl3ЕШЁЃ
ЂВЖрТБЛЏЮя
гааЉН№ЪєТБЛЏЮяФмгыТБЫиЕЅжЪЛђТБЫиЛЅЛЏЮяЗЂЩњМгКЯзїгУЃЌЩњГЩЕФЛЏКЯЮяГЦЮЊЖрТБЛЏЮяЁЃР§ШчЃКKI3ЃЌKICl2ЃЌKI2ClЃЌKIBrClЕШЁЃ
КЌга3ИіТБдзгЕФЖрТБЛЏЮявѕРызгЕФПеМфЙЙаЭМИКѕЖМЪЧжБЯпаЭЕФЁЃШчТБдзгВЛЭЌЪБЃЌдђАыОЖНЯДѓЕФТБдзгЮЛгкжаМфЃЌЖјАыОЖНЯаЁЕФТБдзгЮЛгкСНВрЁЃ
I2дкКЌгаI-ЕФШмвКжаШмНтЖШБШдкДПЫЎжаШмНтЖШДѓЕУЖрЃЌгыЩњГЩЖрТБЛЏЮягаЙиЃЌМДЗЂЩњШчЯТМгКЯЗДгІЃК
KI + I2 Ёњ KI3
11.2.5 ТБЫиЕФКЌбѕЛЏКЯЮя
ЂБТБЫиЕФКЌбѕЛЏКЯЮяИХЪі
ТБЫиЕФКЌбѕЛЏКЯЮягабѕЛЏЮяЁЂЧтбѕЛЏЮя(КЌбѕЫс)ЁЂКЌбѕЫсбЮЃЌЦфЮШЖЈадвРДЮдіЧПЁЃ
ЂВИїРрТБЫиКЌбѕЫсИљЕФНсЙЙ
ЖдгкШЮКЮРраЭЕФТБЫиКЌбѕЫсИљЃЌ ![]() ,МлВуЕчзгЖдЕФПеМфЙЙаЭЮЊЫФУцЬхЙЙаЭЃЌМДТБдзгXЖМВЩгУsp3дгЛЏЃЌТБдзггУsp3дгЛЏЙьЕРгыбѕдзгOГЩМќЁЃ
,МлВуЕчзгЖдЕФПеМфЙЙаЭЮЊЫФУцЬхЙЙаЭЃЌМДТБдзгXЖМВЩгУsp3дгЛЏЃЌТБдзггУsp3дгЛЏЙьЕРгыбѕдзгOГЩМќЁЃ
![]()
![]()
![]()
![]()
ТБдзгбѕЛЏжЕ +1 +3 +5 +7
УћГЦ ДЮТБЫсИљ бЧТБЫсИљ ТБЫсИљ ИпТБЫсИљ
РызгПеМфЙЙаЭ жБЯпаЭ VзжаЭ Ш§НЧзЖ ЫФУцЬх
ПеМфЙЙаЭ


ФЃаЭ 
![]()

ЕчзгНсЙЙЪН ![]()
![]()
![]()

ЂГДЮТБЫсМАЦфбЮ
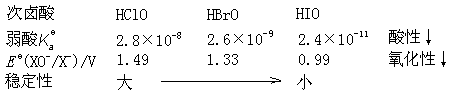
ДЮТШЫсКмВЛЮШЖЈЃЌжЛФмДцдкгкЯЁШмвКжаЃЌЖјВЛФмжЦЕУХЈЫсЁЃДЮТШЫсМћЙтЪмШШОљВЛЮШЖЈЁЃ
![]()
![]()
ДЮфхЫсЁЂДЮЕтЫсЮШЖЈадИќВюЁЃ
ДЮТШЫсЪЧКмЧПЕФбѕЛЏМСЁЃТШЦјЕФЦЏАззїгУОЭЪЧгЩгкЫќгыЫЎзїгУЖјЩњГЩДЮТШЫсЕФдЕЙЪЃЌЫљвдЭъШЋИЩдяЕФТШЦјУЛгаЦЏАззїгУЁЃ
АбТШЦјЭЈШыРфЕФМюШмвКжаЃЌБуЩњГЩДЮТШЫсбЮЁЃ
Cl2 + NaOH Ёњ NaClO + NaCl + H2O
ЦЏАзЗлЪЧгУТШЦјгыЯћЪЏЛвзїгУЖјжЦЕУЕФЃЌЪЧДЮТШЫсИЦЁЂТШЛЏИЦКЭЧтбѕЛЏИЦЕФЛьКЯЮяЁЃ
2Cl2 + 3Ca(OH)2 Ёњ Ca(ClO)2 + CaCl2ЁЄCa(OH)2ЁЄH2O + H2O
фхКЭРфЕФМюШмвКзїгУФмЩњГЩДЮфхЫсбЮЁЃNaBrOдкЗжЮіЛЏбЇЩЯГЃгУзїбѕЛЏМСЁЃДЮЕтЫсЕФЮШЖЈадМЋВюЃЌЫљвдЃЌЕтгыМюШмвКЗДгІЕУВЛЕНДЮЕтЫсбЮЁЃ
*ЂДбЧТБЫсМАЦфбЮ
ФПЧАвбЛёЕУЕФбЧТБЫсМАЦфбЮЪЧбЧТШЫсМАЦфбЮЁЃЗДгІЮЊЃК
2ClO2 + H2O Ёњ HClO2 + HClO3
Ba(ClO)2 + H2SO4 Ёњ 2HClO2 + BaSO4
ЖўбѕЛЏТШгыЙ§бѕЛЏЮяЗДгІЪБЃЌПЩЕУЕНбЧТШЫсбЮКЭбѕЁЃ
2ClO2 + Na2O2 Ёњ 2NaClO2 + O2
2ClO2 + BaO2 Ёњ Ba(ClO2)2 + O2
бЧТШЫсбЮЫфБШбЧТШЫсЮШЖЈЃЌЕЋМгШШЛђЧУЛїЙЬЬхбЧТШЫсбЮЪБЃЌСЂМДЗЂЩњБЌеЈЃЌЗжНтГЩЮЊТШЫсбЮКЭбѕЛЏЮяЁЃбЧТШЫсбЮЕФЫЎШмвКНЯЮШЖЈЃЌОпгаЧПбѕЛЏадЃЌПЩзїЦЏАзМСЁЃ
ЂЕТБЫсМАЦфбЮ

ТШЫсЪЧЧПЫсЃЌНіДцдкгкЫЎШмвКжаЁЃТШЫсвВЪЧЧПбѕЛЏМСЃЌР§Шч
2HClO3 + I2 Ёњ 2HIO3 + Cl2
HClO3 + 5HCl Ёњ 3Cl2 + 2H2O
живЊЕФТШЫсбЮгаТШЫсМиКЭТШЫсФЦЁЃKClO3ЪмШШЗжНтЃЌвђЬѕМўВЛЭЌЖјВњЮяВЛЭЌЁЃ
![]()
4KClO3Ёњ3KClO4+KCl]
ЙЬЬхТШЫсМиKClO3ЪЧЧПбѕЛЏМСЃЌгыИїжжвзШМЮяЛьКЯКѓЃЌОзВЛїЛсв§Ц№БЌеЈЦ№Л№ЁЃвђДЫЃЌKClO3ЖргУРДжЦдьЛ№ВёКЭбцЛ№ЕШЁЃ
KClO3КЭC11H22O11ЛьКЯЮяЕФЛ№бц KClO3ЁЊЛ№ВёЭЗжаЕФбѕЛЏМС
ГЃгУТШЫсБЕгыЯЁСђЫсзїгУжЦШЁТШЫсЃК Ba(ClO3)2 + H2SO4 Ёњ BaSO4 + 2HClO3
фхЫсКЭЕтЫсЕФжЦБИЃЌЭЈГЃгУбЁдёЪЪЕБЕФбѕЛЏМСЖјЛёЕУЁЃР§ШчЃК
Br2 + 5Cl2 + 6H2OЁњ 2HBrO3 + 10HCl
I2 + 5Cl2 + 6H2OЁњ 2HIO3 + 10HCl
3I2 + 10HNO3 Ёњ 6HIO3 + 10NO + 2H2O
ЂЖИпТБЫсМАЦфбЮ
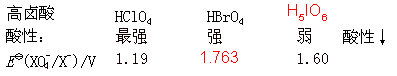
ИпТБЫсЖМЪЧЧПбѕЛЏМСЃЌОљвбЛёЕУДПЮяжЪЃЌЮШЖЈадКУЁЃР§ШчЃК
![]()
ИпТШЫсЪЧзюЧПЕФЮоЛњКЌбѕЫсЁЃЮоЫЎЕФИпТШЫсЪЧЮоЩЋвКЬхЁЃHClO4ЕФЯЁШмвКБШНЯЮШЖЈЃЌЕЋХЈHClO4ВЛЮШЖЈЃЌЪмШШЗжНтЁЃ
4HClO4 Ёњ 2Cl2 + 7O2 + 2H2O
ХЈHClO4ЪЧЧПбѕЛЏМСЃЌгыгаЛњЮяНгДЅЛсв§Ц№БЌеЈЃЌЫљвджќДцЪББиаыдЖРыЛЙдадЮяжЪЁЃЮёБизЂвтАВШЋЃЁИпТШЫсдкИжЬњЗжЮіжаГЃгУРДШмНтПѓбљЁЃ
ИпТШЫсЕФбЮЖрвзШмгкЫЎЃЌЕЋK+ЃЌ ![]() ЃЌCs+ЃЌRb+ЕФИпТШЫсбЮШмНтЖШЖМКмаЁЁЃгааЉИпТШЫсбЮвзЮќЪЊЃЌШчMg(ClO4)2КЭBa(ClO4)2ПЩзїИЩдяМСЁЃKClO4ГЃгУзїжЦдьеЈвЉЁЃNH4ClO4ЪЧЯжДњЛ№М§ЭЦНјМСЕФжївЊГЩЗжЁЃ
ЃЌCs+ЃЌRb+ЕФИпТШЫсбЮШмНтЖШЖМКмаЁЁЃгааЉИпТШЫсбЮвзЮќЪЊЃЌШчMg(ClO4)2КЭBa(ClO4)2ПЩзїИЩдяМСЁЃKClO4ГЃгУзїжЦдьеЈвЉЁЃNH4ClO4ЪЧЯжДњЛ№М§ЭЦНјМСЕФжївЊГЩЗжЁЃ
ЂЗТШЕФИїжжКЌбѕЫсМАЦфбЮЕФаджЪЕФвЛАуЙцТЩ

Ёь11.3ЁЁбѕзхдЊЫи
11.3.1 бѕзхдЊЫиИХЪі
бѕзхдЊЫиЪЧжмЦкЯЕЕкЂіAзхдЊЫиЃЌАќРЈбѕ(O)ЁЂСђ(S)ЁЂЮј(Se)ЁЂэк(Te)ЁЂюЧ(Po)ЮхжждЊЫиЃЌЦфМлЕчзгЙЙаЭЮЊЃКns2np4ЁЃ
дЊЫиЗћКХ | O | S | Se | Te | Po |
ЕЅжЪаджЪ | ЕфаЭЗЧН№Ъє | зМН№Ъє | ЗХЩфадН№Ъє | ||
ДцдкаЮЪН | ЕЅжЪЛђПѓЮя | ЙВЩњгкжиН№ЪєЕФСђЛЏЮяжа | |||
ЕЅжЪЛЏбЇЛюад | гЩДѓЕНаЁ | ||||
БОНкЕФЬжТлжиЕуЪЧбѕЁЂСђМАЦфЛЏКЯЮяЁЃЮјЁЂэкЕФЕЅжЪМћЯТЭМЃК
![]()
![]()
11.3.2 бѕМАЦфЛЏКЯЮя
ЂБбѕ
бѕЪЧЕиПЧжаЗжВМзюЙуЗКЕФдЊЫиЃЌЦфЗсЖШОгИїжждЊЫижЎЪзЃЌЦфжЪСПдМеМЕиПЧЕФвЛАыЁЃдкКЃбѓжабѕвдЫЎЕФаЮЪНДцдкЃЌдкДѓЦјжабѕвдЕЅжЪзДЬЌДцдкЃЌдкбвЪЏКЭЭСШРжабѕвдЙшЫсбЮЁЂбѕЛЏЮяМАЦфЫќКЌбѕвѕРызгЕФаЮЪНДцдкЁЃ
бѕЕФЕчзгХХВМЪНЮЊЃК ![]()
ЦфНсЙЙЪНЮЊЃК ![]() ,ОпгаЫГДХадЁЃ
,ОпгаЫГДХадЁЃ
ГЃЮТЯТбѕЦјжЛФмНЋФГаЉЛЙдадЕФЮяжЪ(ШчNOЃЌSnCl2ЃЌH2SO3ЕШ)бѕЛЏЁЃдкМгШШЬѕМўЯТЃЌГ§ТБЫиЁЂЩйЪ§ЙѓН№Ъє(ШчAgЁЂPtЕШ)вдМАЯЁгаЦјЬхЭтЃЌбѕЦјМИКѕФмгыЫљгадЊЫижБНгЛЏКЯГЩЯргІЕФбѕЛЏЮяЁЃ
ЂВГєбѕ
ГєбѕO3ЪЧбѕЦјO2ЕФЭЌЫивьаЮЬхЁЃГєбѕдкЕиУцИННќЕФДѓЦјВужаКЌСПМЋЩйЃЌЖјдкДѓЦјВуЕФзюЩЯВуЃЌгЩгкЬЋбєЖдДѓЦјжаЕФбѕЦјЕФЧПСвЗјЩфзїгУЃЌаЮГЩСЫвЛВуГєбѕВуЁЃГєбѕВуФмЮќЪеЬЋбєЙтЕФзЯЭтЗјЩфЃЌГЩЮЊБЃЛЄЕиЧђЩЯЩњУќУтЪмЬЋбєНЋЗјЩфЕФЬьШЛЦСеЯЁЃ
 ГєбѕЗжзгЕФЙЙаЭЮЊVаЭЃЌШчЭМЫљЪОЃК
ГєбѕЗжзгЕФЙЙаЭЮЊVаЭЃЌШчЭМЫљЪОЃК
жааФбѕдзгвд2Иіsp2дгЛЏЙьЕРгыСэЭтСНИібѕдзгаЮГЩІвМќЃЌЕкШ§Иіsp2дгЛЏЙьЕРБЛЙТЖдЕчзгЫљеМгаЁЃДЫЭтЃЌжааФбѕдзгЕФЮДВЮгыдгЛЏЕФpЙьЕРЩЯгавЛЖдЙТЖдЕчзгЃЌСНЖЫЕФбѕдзггыЦфЦНааЕФpЙьЕРЩЯИїгавЛИіЕчзгЃЌЫќУЧжЎМфаЮГЩДЙжБгкЗжзгЦНУцЕФШ§жааФЫФЕчзгДѓІаМќЃЌгУ ![]() БэЪОЁЃ
БэЪОЁЃ
ГєбѕЗжзгжаМќНЧЮЊ117Ёу,ЗжзгЕФХММЋОиІЬ=1.8ЁС10-3cЁЄmЁЃГєбѕЪЧЮЈвЛЕФМЋадЕЅжЪЁЃ
ГєбѕЪЧЕРЖЩЋЕФЦјЬхЃЌгагуаШЮЖЁЃГєбѕМЋВЛЮШЖЈЃЌдкГЃЮТЯТЛКТ§ЗжНтЃК
O3(g) Ёњ 3O2(g)
ЖўбѕЛЏУЬЕФДцдкПЩМгЫйГєбѕЕФЗжНтЁЃГєбѕЕФСэвЛИіживЊаджЪОЭЪЧЫќЕФЧПбѕЛЏадЃЌР§ШчЃК
O3 + 2I- + 2H+ Ёњ I2 + O2 + H2O
етИіЗДгІгУгкВтЖЈГєбѕЕФКЌСПЁЃ
РћгУГєбѕбѕЛЏадКЭВЛвзЕМжТЖўДЮЮлШОЕФгХЕуЃЌГєбѕПЩгУзїЯћЖОМСЃЌгУРДОЛЛЏЗЯЦјЁЂЗЯЫЎЁЃ
ЂГЙ§бѕЛЏЧт
Й§бѕЛЏЧтH2O2ЕФЗжзгНсЙЙЃКH2O2ЕФЗжзгНсЙЙШчЭМЫљЪОЃЌдкH2O2ЕФЗжзгжагавЛИіЙ§бѕМќЉЄOЉЄOЉЄЃЌСНИібѕдзгЖМвдsp3дгЛЏЙьЕРГЩМќЃЌГ§ЯрЛЅСЌНгГЩOЉЄOМќЭтЃЌЛЙИїгывЛИіЧтдзгЯрСЌЁЃ
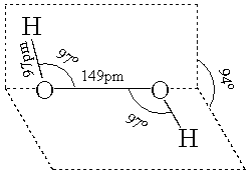
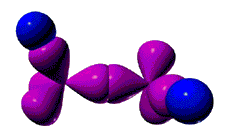
H2O2ЕФаджЪЃКИпДПЖШЕФH2O2дкЕЭЮТЯТБШНЯЮШЖЈЃЌЗжНтзїгУБШНЯЦНЮШЁЃЕБМгШШЕН426KвдЩЯЃЌЗЂЩњБЌеЈадЗжНтЃК
2H2O2(l) Ёњ 2H2O + O2(g)
вђДЫЃЌH2O2гІжќДцдкзиЩЋЦПжаЃЌжУгквѕСЙДІЁЃ
H2O2ОпгаШѕЫсадКЭбѕЛЏЛЙдадЁЃР§ШчЃК
ШѕЫсадЃК H2O2 + Ba(OH)2 Ёњ BaO2 + 2H2O
ЧПбѕЛЏадЃК | ЫсадЬѕМўЃК2Fe2+ + H2O2 + 2H+ Ёњ 2Fe3+ + 2H2O |
МюадЬѕМўЃК2[Cr(OH)4]- + 3H2O2 + 2OH- Ёњ 2[CrO4]2- + 8H2O |
Й§бѕЛЏЧтЛЙПЩНЋКкЩЋЕФPbSбѕЛЏЮЊАзЩЋЕФPbSO4ЃК
PbS + 4H2O2 Ёњ PbSO4 + 4H2O
жЛгаЕБH2O2гыЧПбѕЛЏМСзїгУЪБВХЯдЪОГіЦфЛЙдадЁЃ
2KMnO4 + 5H2O2 + 3H2SO4 Ёњ 2MnSO4 + 5O2 + K2SO4 + 8H2O
H2O2 + Cl2 Ёњ 2HCl + O2
Й§бѕЛЏЧтвВЪЧвЛжжВЛдьГЩЖўДЮЮлШОЕФбѕЛЏМСЃЌЫљвдГЃгУзїЩБОњМСЁЂЦЏАзМСЕШЁЃзЂвтЃКХЈЖШЩдДѓЕФЫЋбѕЫЎЛсзЦЩЫЦЄЗєЃЌЪЙгУЪБгІИёЭтаЁаФ!
11.3.3 СђМАЦфЛЏКЯЮя
ЂБЕЅжЪСђ
ЕЅжЪСђЫзГЦСђЛЧЃЌЪЧЗжзгОЇЬхЃЌКмЫЩДрЃЌВЛШмгкЫЎЁЃСђгаМИжжЭЌЫивьаЮЬхЃЌаБЗНСђ(СтаЮСђ)ЁЂЕЅаБСђЁЂЕЏадСђЕШЁЃЬьШЛСђвЛАуЪЧаБЗНСђЁЃ
| аБЗНСђ | ЕЅаБСђ | ЕЏадСђ |
УмЖШ/gЁЄcm-1 | 2.06 | 1.99 |
|
беЩЋ | ЛЦЩЋ | ЧГЛЦ |
|
ЮШЖЈЬѕМў | ЃМ94.5Ёц | 94.5ЁцЁЋ115Ёц | 190ЁцЕФШлШкСђгУРфЫЎЫйРф |
ЭтЙл |
|
|
|

аБЗНСђКЭЕЅжЪСђЕФЗжзгЖМЪЧгЩ8ИіСђдзгзщГЩЕФЛЗзДНсЙЙЃЌЯёЛЪЙквЛбљЃЌШчЭМЫљЪОЁЃдкS8ЗжзгжаЃЌУПИіСђдзгИївдsp3дгЛЏЙьЕРжаЕФСНИіЙьЕРгыЯрСкЕФСНИіСђдзгаЮГЩІвМќЃЌЖјsp3дгЛЏЙьЕРжаЕФСэСНИідђИїгавЛЖдЙТЖдЕчзгЁЃЕЅжЪСђгыаБЗНСђЕФВЛЭЌдкгкЫФУцЬхжаS8ЗжзгХХСаВЛЭЌЁЃ
![]() ЕЅжЪСђЪЧЗжзгОЇЬхЃЌШлЕуЕЭЃЌВЛШмгкЫЎЖјвзШмгкЖўСђЛЏЬМЃЌЫФТШЛЏЬМЕШЗЧМЋадШмМСЁЃ
ЕЅжЪСђЪЧЗжзгОЇЬхЃЌШлЕуЕЭЃЌВЛШмгкЫЎЖјвзШмгкЖўСђЛЏЬМЃЌЫФТШЛЏЬМЕШЗЧМЋадШмМСЁЃ
ЕЅжЪСђМгШШШлЛЏКѓЃЌЮТЖШЩ§жС160ЁцЃЌS8ЛЗПЊЪМЖЯПЊЃЌВЂОлКЯГЩжаГЄСДДѓЗжзгЃЌвђЖјвКЬхбеЩЋБфАЕЃЌеГЖШЯджјдіДѓЁЃЕБЮТЖШДяЕН190ЁцзѓгвЃЌЕЙШыРфЫЎжабИЫйРфШДЃЌПЩвдЕУЕНЕЏадСђЁЃДЫЪБЃЌгЩгкжшРфЃЌГЄСДзДСђЗжзгРДВЛМАГЩЛЗЃЌШдвдНЪНсЕФГЄСДДцдкгкЙЬЬхжаЃЌвђЖјЙЬЬхОпгаЕЏадЁЃ
СђЕФЛЏбЇаджЪБШНЯЛюЦУЃЌФмгыаэЖрН№ЪєжБНгЛЏКЯГЩЯргІЕФСђЛЏЮяЃЌвВФмгыЧтЁЂбѕЁЂТБЫи(Г§ЕтЭт)ЁЂЬМСзЕШжБНгзїгУЩњГЩЯргІЕФЙВМлЛЏКЯЮяЁЃ
СђгыбѕЛЏадЫсЕФзїгУЃК | S + 2HNO3 Ёњ H2SO4 + 2NO(g) |
S + 2H2SO4(ХЈ) Ёњ 3SO2(g) + 2H2 | |
СђдкМюжаЕФЦчЛЏЃК |
|
|
ЂВСђЛЏЧтКЭСђЛЏЮя
ЂХСђЛЏЧт
СђЛЏЧтH2SЗжзгЕФЙЙаЭгыЫЎЗжзгЯрЫЦЃЌвВГЪ"V"зжаЮЃЌЕБЦфЗжзгМЋадБШH2OШѕЁЃ
ЭЈГЃгУН№ЪєСђЛЏЮяКЭЗЧбѕЛЏадЫсжЦБИСђЛЏЧтЃК
FeS + 2HCl Ёњ H2S + FeCl2
вВПЩгУСђДњввѕЃАЗЫЎШмвКМгШШЫЎНтЕФЗНЗЈжЦШЁЁЃ
CH3CSNH2 + 2H2O Ёњ CH3COONH4 + H2S
СђЛЏЧтЪЧЮоЩЋгаИЏЕАЮЖЕФОчЖОЦјЬхЁЃH2SжаЖОЪЧгЩгкЫќФмгыбЊКьЫижаFe2+зїгУЩњГЩFeSГСЕэЃЌвђЖјЪЙFe2+ЪЇШЅдРДе§ГЃЕФЩњРэзїгУЁЃСђЛЏЧтЮЂШмгкЫЎЃЌЦфЫЎШмвКЮЊЖўдЊШѕЫсЁЃ
СђЛЏЧтзюживЊЕФаджЪОЭЪЧЫќЕФЧПЛЙдадЁЃ
Ёя БЛПеЦјбѕЛЏ
![]()
![]()
ЫљвдСђЛЏЧтЫЎШмвКдкПеЦјжаЗХжУЛсж№НЅБфЛьзЧЁЃ
Ёя гыжаЕШЧПЖШбѕЛЏМСзїгУ
H2S + 2Fe3+ Ёњ S + 3Fe2+ + 2H+ (зЂвтЃКЗДгІВЛЩњГЩFe2S3ЛђFeS)
H2S + X2 Ёњ S + 2X- + 2H+ (X=Cl,Br,I)
Ёя гыЧПЖШбѕЛЏМСЗДгІ
![]()
![]()
ЂЦН№ЪєСђЛЏЮя
беЩЋЃКН№ЪєСђЛЏЮяДѓЖрЪ§ЮЊКкЩЋЃЌЩйЪ§ашЬиЪтМЧвфЁЃР§ШчЃКSnSзиЩЋ,SnS2ЛЦЩЋ,As2S3ЛЦЩЋ,As2S5ЛЦЩЋ,Sb2S3ГШЩЋ,As2S5ГШЩЋ,MnSШтЩЋ,ZnSАзЩЋ,CdSЛЦЩЋЁЃ
ЫЎНтадЃКН№ЪєСђЛЏЮяЮоТлЪЧЮЂШмЛЙЪЧвзШмЃЌЖМЛсЗЂЩњЫЎНтЗДгІЃЌМДЪЙЪЧФбШмН№ЪєСђЛЏЮяЃЌЦфШмНтВПЗжвВ
ЛсЗЂЩњЫЎНтЁЃР§ШчЃКNa2SЁЂ(NH4S)ЫЎШмвКвђЫЎНтЖјГЪМюад;Cr2S3ЁЂAl2S3гіЫЎЗЂЩњЧПСвЫЎНтЃК
2M3+ + 3S2- + 6H2O Ёњ 2M(OH)3 + 3H2S (M=Al,Cr)
ШмНтадЃК ![]() КЭМюН№ЪєСђЛЏЮяМАBaSвзШмгкЫЎЃЌЦфЫћМюЭСН№ЪєСђЛЏЮяЮЂШмгкЫЎ(BeSФбШм)ЁЃГ§ДЫжЎЭтЃЌДѓЖрЪ§Н№ЪєСђЛЏЮяФбШмгкЫЎЁЃФбШмгкЫЎЕФН№ЪєСђЛЏЮядкВЛЭЌЕФЫсЁЂМюжаШмНтГЬЖШвВгаВювьЃЌРћгУетаЉВювьЮвУЧПЩНЋН№ЪєРызгНјааЗжРыЁЃ
КЭМюН№ЪєСђЛЏЮяМАBaSвзШмгкЫЎЃЌЦфЫћМюЭСН№ЪєСђЛЏЮяЮЂШмгкЫЎ(BeSФбШм)ЁЃГ§ДЫжЎЭтЃЌДѓЖрЪ§Н№ЪєСђЛЏЮяФбШмгкЫЎЁЃФбШмгкЫЎЕФН№ЪєСђЛЏЮядкВЛЭЌЕФЫсЁЂМюжаШмНтГЬЖШвВгаВювьЃЌРћгУетаЉВювьЮвУЧПЩНЋН№ЪєРызгНјааЗжРыЁЃ
Ёя ЯЁЫсШмНтадСђЛЏЮя:MnSЁЂFeSЁЂCoSЁЂNiSЁЂZnSЁЃ
MS + 2H+ Ёњ M2+ + H2S(g)
Ёя ХЈбЮЫсХфЮЛШмНт
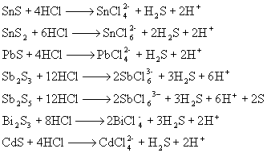
Ёя ЯѕЫсбѕЛЏШмНтЃК
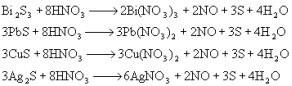
Ёя ЭѕЫЎШмНтЃК
![]()
Ёя МюШм(гУNaOHЛђNa2S)
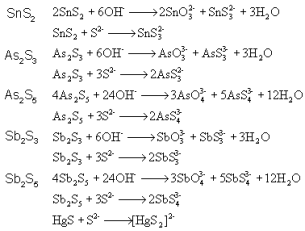
Ёя бѕЛЏМюШм(Na2S2)
ЂГЖрСђЛЏЮя
дкПЩШмадСђЛЏЮяЕФШмвКжаМгШыСђЗлЪБЃЌСђШмНтЖјЩњГЩЯргІЕФЖрСђЛЏЮяЃЌР§ШчЃК
(NH4)2S + (x-1)S Ёњ (NH4)2Sx
ЫцзХСђдзгЪ§ФПxЕФдіМгЃЌШмвКбеЩЋгЩЛЦЁњГШКьЁњКьЁЃЫљвдЭЈГЃЫљЕУЕФЖрСђЛЏЮяЪЧКЌгаВЛЭЌЪ§ФПСђдзгЕФИїжжЖрСђЛЏЮяЕФЛьКЯЮяЁЃ
ЖрСђРызг ![]() ОпгаСДЪННсЙЙЃК
ОпгаСДЪННсЙЙЃК ![]()
ЕБx=2ЪБЃЌЖрСђЛЏЮяГЦЮЊЙ§СђЛЏЮя(ЭЌЙ§бѕЛЏЮя)ЁЃ
Й§СђЛЏЧтH2S2гыЙ§бѕЛЏЧтЕФНсЙЙЯрЫЦЁЃ
ЖрСђЛЏЮяЕФживЊаджЪЃК
Ёя гіЫсВЛЮШЖЈ
![]()
Ёя ЛЙдад(ДЮвЊ)
3FeS2 + 8O2 Ёњ Fe3O4 + 6SO3
Ёя бѕЛЏад(жївЊ)
![]()
ЫљвдЖрСђЛЏЮягУзїКёЦЄЕФГ§УЋМСЃЌЙћФОЁЂУоЛЈЕФЩБГцМСЁЃ
ЂДЖўбѕЛЏСђЁЂбЧСђЫсМАЦфбЮ
ЖўбѕЛЏСђжЦБИЃКЙЄвЕЩЯРћгУБКЩеСђЛЏЮяПѓжЦШЁSO2ЃК
3FeS2 + 8O2 Ёњ Fe3O4 + 6SO2
ЪЕбщЪвжаГЃгУбЧСђЫсбЮгыЫсзїгУжЦШЁЩйСПЖўбѕЛЏСђЃЌвВПЩгУХЈСђЫсгыЭЙВШШжЦШЁЖўбѕЛЏСђЁЃ
ЖўбѕЛЏСђЕФНсЙЙЃКЦјЬЌЕФSO2ЕФЗжзгЙЙаЭЮЊVаЮЃЌШчЭМЫљЪОЁЃSO2ЗжзгжаЃЌСђдзгвдСНИіsp2дгЛЏЙьЕРЗжБ№гыСНИібѕдзгаЮГЩІвМќЃЌЖјСэвЛИіsp2дгЛЏЙьЕРЩЯдђБЃСє1ЖдЙТЖдЕчзгЃЌСђдзгЕФЮДВЮгыдгЛЏЕФpЙьЕРЩЯЕФ2ИіЕчзггыСНИібѕдзгЕФЮДГЩЖдpЕчзгаЮГЩШ§жааФЫФЕчзгДѓІа Мќ
Мќ ![]() ЁЃ
ЁЃ
ЖўбѕЛЏСђКЭбЧСђЫсЕФаджЪЃКSO2ЪЧЮоЩЋЁЂОпгаЧПСвДЬМЄадЦјЮЖЕФЦјЬхЁЃвКЬЌSO2ФмЙЙНтРыЃЌЪЧвЛжжСМКУЕФЗЧЫЎШмМСЁЃ
![]()
SO2ЕФЦћЛЏьЪКмДѓЃЌвКЬЌSO2ПЩгУзїжЦРфМСЁЃ
SO2взШмгкЫЎЃЌЩњГЩКмВЛЮШЖЈЕФбЧСђЫсH2SO3ЁЃ
Ёя H2SO3ЪЧЖўдЊжаЧПЫсЃЌ ![]() ЃЌ
ЃЌ ![]() ЃЌH2SO3жЛДцдкгкЫЎШмвКжаЃЌЙтЦзЪдбщжЄУїSO2дкЫЎШмвКжаЕФзДЬЌЛљБОЩЯЪЧSO2ЁЄH2OЁЃ
ЃЌH2SO3жЛДцдкгкЫЎШмвКжаЃЌЙтЦзЪдбщжЄУїSO2дкЫЎШмвКжаЕФзДЬЌЛљБОЩЯЪЧSO2ЁЄH2OЁЃ
Ёя H2SO3ЪЧНЯЧПЕФЛЙдМСЃЌР§ШчЃК
ЫљвдПеЦјжаГЄЦкЗХжУЕФбЧСђЫсЛђбЧСђЫсбЮЛсБЛПеЦјжаЕФбѕбѕЛЏЖјЪЇШЅЛЙдадЁЃ
Ёя бѕЛЏадЃКH2SO3гыЧПЛЙдМСЗДгІЪБВХБэЯжГібѕЛЏадЁЃ
H2SO3 + H2S Ёњ 3S + 3H2O
Ёя ЦЏАззїгУSO2ЛђH2SO3ПЩгыФГаЉгаЛњЮяЗЂЩњМгКЯзїгУЃЌЖјЪЙгаЛњЮяЭЫЩЋЁЃР§ШчЫќУЧФмЪЙЦЗКьШмвКЭЫЩЋЁЃ
SO2жївЊгУгкЩњВњСђЫсКЭбЧСђЫсбЮЃЌЛЙДѓСПгУгкЩњВњКЯГЩЯДЕгМСЁЂЪГЦЗЗРИЏМСЁЂзЁЫљКЭгУОпЯћЖОМСЕШЁЃ
ЂЕШ§бѕЛЏСђЁЂСђЫсМАЦфбЮ
ЂХШ§бѕЛЏСђ
дкМгШШКЭДпЛЏМСДцдкЯТПЩНЋSO2бѕЛЏГЩSO3ЁЃ
НсЙЙЃКЦјЬЌSO3ЮЊЕЅЗжзгЃЌЦфЗжзгНсЙЙЮЊЦНУцШ§НЧаЮЁЃ
дкSO3ЗжзгжаЃЌСђдзгвдsp2дгЛЏЙьЕРгы3ИібѕдзгаЮГЩ3ИіІвМќЃЌДЫЭтЃЌЛЙвдpd2дгЛЏЙьЕРгы3ИібѕдзгаЮГЩДЙжБгкЗжзгЦНУцЕФЫФжааФСљЕчзгДѓІАМќ ![]() ЁЃМДЃК
ЁЃМДЃК

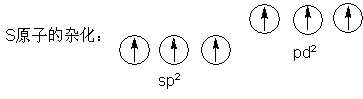
дкДѓІАМќжагаШ§ИіЕчзгдРДЪєгкСђдзгЃЌЖјСэЭтШ§ИіЕчзгдРДЗжБ№ЪєгкШ§ИібѕдзгЁЃдкЙЬЬЌSO3жаЃЌСђдзгЖМВЩгУsp3дгЛЏЙьЕРГЩМќЁЃЙЬЬхШ§бѕЛЏСђгаМИжжОлКЯОЇаЭЁЃдкВЛЭЌРраЭSO3ЙЬЬхжаЃЌSO3ЗжзгЕФХХСаЗНЪНВЛЭЌЁЃІУаЭОЇЬхЮЊШ§ОлЗжзгЃЌІТаЭОЇЬхЮЊSO3дзгЭХЛЅЯрСЌНгГЩЕФГЄСДЃЌгыЪЏУоНсЙЙРрЫЦЃЌІСаЭОЇЬхвВОпгаРрЫЦЪЏУоЕФНсЙЙЁЃ
аджЪЃКДПШ§бѕЛЏСђЪЧвЛжжЮоЩЋЁЂвзЛгЗЂЕФЙЬЬхЃЌЦфШлЕуЮЊ16.8Ёц,ЗаЕуЮЊ44.8ЁцЁЃSO3ОпгаКмЧПЕФбѕЛЏадЁЃР§ШчЃЌЕБСзКЭЫќНгДЅЪБЛсШМЩеЁЃИпЮТЪБSO3ЕФбѕЛЏадИќЮЊЯджјЃЌЫќФмбѕЛЏKI,HBrКЭFe,ZnЕШН№ЪєЁЃ
Ш§бѕЛЏСђМЋвзгыЫЎЛЏКЯЩњГЩСђЫсЃЌЭЌЪБЗХГіДѓСПЕФШШЃК
![]()
вђДЫЃЌSO3дкГБЪЊЕФПеЦјжаГЪЮэзДЁЃ
ЂЦСђЫс
НсЙЙЃКдкСђЫсЗжзгжаЃЌСђдзгВЩШЁsp3дгЛЏЙьЕРгыЫФИібѕдзгЕФ2ИіаЮГЩ2ИіІвМќЃЛСэСНИідђНгЪмСђЕФЕчзгЖдЗжБ№аЮГЩІвХфМќЃЛгыДЫЭЌЪБЃЌСђдзгЕФПеЕФ3dЙьЕРгыСНИіВЛдкOHЛљжаЕФбѕдзгЕФ2pЙьЕРЖдГЦадЦЅХфЃЌЯрЛЅжиЕўЃЌЗДЙ§РДНгЪмРДзд2ИібѕдзгЕФЙТЖдЕчзгЃЌДгЖјаЮГЩСЫИНМгЕФ(pЉЄd)ІаЗДРЁХфМќЁЃ
СђЫсОЇЬхГЪЯжВЈЮЦаЮВузДНсЙЙЁЃУПИіСђбѕЫФУцЬх(SO4дзгЭХ)ЭЈЙ§ЧтМќгыЦфЫќ4ИіSO4МЏЭХСЌНгЁЃ
аджЪЃКСђЫсЪЧЖўдЊЧПЫсЃЌНЯЮШЖЈЁЃХЈСђЫсОпгаЧПЮќЫЎадКЭЧПбѕЛЏадЁЃ
Ёя ЧПЮќЫЎадЃКХЈСђЫсЕФЮќЫЎадКмЧПЃЌЫќГ§СЫПЩзїИЩдяМСЭтЃЌЫќЛЙФмДгЯЫЮЌКЭЬЧжаАДH:OЮЊ2:1ЕФБШР§ЖсШЁH2O,ЪЙЦфЬМЛЏЁЃ
C12H22O11 Ёњ 12C + 11H2O
Ёя ЧПбѕЛЏадЃКХЈСђЫсПЩвдбѕЛЏаэЖрН№ЪєКЭФГаЉЗЧН№ЪєЁЃ
гыЛюЦУН№ЪєЃК 3Zn + 4H2SO4(ХЈ) Ёњ 3ZnSO4 + S + H2O
4Zn + 5H2SO4(ХЈ) Ёњ4ZnSO4 + H2S + H2O
гыВЛЛюЦУН№ЪєЃК Cu + 2H2SO4(ХЈ) Ёњ CnSO4 + SO2 + 2H2O
гыЗЧН№ЪєЃК C+ 2H2SO4(ХЈ) Ёњ CO2 + 2SO2 + 2H2O
2P+ 5H2SO4(ХЈ) Ёњ P2O2 + 5SO2 + 5H2O
S+ 2H2SO4(ХЈ) Ёњ 3SO2 + 2H2O
РфЕФХЈСђЫс(70%вдЩЯ)ФмЪЙЬњЕФБэУцЖлЛЏЃЌЩњГЩвЛВужТУмЕФБЃЛЄФЄЃЌзшжЙСђЫсгыЬњЕФБэУцМЬајзїгУЁЃвђДЫПЩвдгУИжЙожќзАКЭдЫЪфХЈСђЫс(80%ЁЋ90%)ЁЃ
ЂЧСђЫсбЮ
СђЫсФмаЮГЩе§бЮКЭЫсЪНбЮЁЃ ![]() ЕФНсЙЙЮЊЫФУцЬхЁЃДѓЖрЪ§СђЫсбЮвзШмгкЫЎЃЌЕЋPbSO4ЁЂCaSO4ЁЂSrSO4ШмНтЖШКмаЁЁЃBaSO4МИКѕВЛШмгкЫЎЧвВЛШмгкЫсЃЌРћгУBaSO4ЕФетвЛЬиадЃЌПЩвдгУBaCl2ЕШПЩШмадБЕбЮМјЖЈ
ЕФНсЙЙЮЊЫФУцЬхЁЃДѓЖрЪ§СђЫсбЮвзШмгкЫЎЃЌЕЋPbSO4ЁЂCaSO4ЁЂSrSO4ШмНтЖШКмаЁЁЃBaSO4МИКѕВЛШмгкЫЎЧвВЛШмгкЫсЃЌРћгУBaSO4ЕФетвЛЬиадЃЌПЩвдгУBaCl2ЕШПЩШмадБЕбЮМјЖЈ ![]() ЕФДц дкЁЃ
ЕФДц дкЁЃ
ЂЖСђЫсЕФЦфЫќКЌбѕЫсМАЦфбЮ
ЂХНЙСђЫсМАЦфбЮ
РфШДЗЂбЬСђЫсПЩвдЮіГіНЙСђЫсH2S2O7,ЮоЩЋОЇЬхЃЌНЙСђЫсЕФНсЙЙЪНЮЊЃК
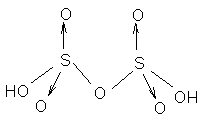
НЙСђЫсЕФЫсадЁЂЮќЫЎадЁЂИЏЪДадБШСђЫсИќЧПЁЃНЙСђЫсЪЧвЛжжЧПбѕЛЏМСЃЌгжЪЧвЛжжСМКУЕФЛЧЛЏМСЃЌЙЄвЕЩЯгУгкжЦдьШОСЯЁЂеЈвЉЁЂКЭЦфЫќгаЛњЛЧЫсЛЏКЯЮяЁЃ
НЙСђЫсбЮПЩгыФГаЉМШВЛШмгыЫЎгжВЛШмгкЫсЕФН№ЪєбѕЛЏЮя(ШчAl2O3ЁЂFe3O4ЁЂTiO2ЕШ)ЙВШлЃЌЩњГЩПЩШмгыЫЎЕФСђЫсбЮЁЃ
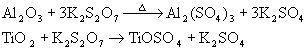
етЪЧЗжЮіЛЏбЇжаДІРэФГаЉЙЬЬхЪдбљЕФвЛжжживЊЗНЗЈЁЃ
ЂЦСђДњСђЫсМАЦфбЮ
СђДњСђЫсH2S2O3ПЩПДзїЪЧСђЫсЗжзгжаЕФвЛИібѕдзгБЛСђдзгЫљШЁДњЕФВњЮяЁЃСђДњСђЫсМЋВЛЮШЖЈЃЌжСНёЩаЮДжЦЕУДПЦЗЁЃ
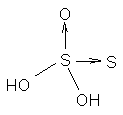
бЧСђЫсбЧгыСђзїгУЩњГЩСђДњСђЫсбЮЁЃР§ШчЃЌНЋСђЗлгыбЧСђЫсФЦвЛПщжѓЗаЃЌПЩжЦЕУСђДњСђЫсФЦЃК
![]()
СэЭтЃЌдкNa2SКЭNa2CO3ЛьКЯШмвК(ЮяжЪЕФСПБШЮЊ2:1)жаЭЈШыSO2вВПЩвджЦЕУNa2S2O3ЃК
2Na2S + Na2CO3 + 4SO2 Ёњ 3Na2S2O3 + CO2
Na2S2O3ЁЄ5H2OЪЧживЊЕФСђДњСђЫсбЮЃЌЫзГЦКЃВЈЛђДѓЫеДђЃЌЪЧЮоЩЋЭИУїЕФОЇЬхЃЌвзШмгкЫЎЃЌЦфЫЎШмвКГЪШѕМюадЁЃ
СђДњСђЫсбЮЕФживЊаджЪШчЯТЃК
Ёя гіЫсЗжНтЃК
![]()
Ёя ЛЙдадЃК
![]()
дкЗФжЏЙЄвЕЩЯгУNa2S2O3зїЭбТШМСЁЃ
![]()
дкЗжЮіЛЏбЇЩЯгУгкЕтСПЗЈЕЮЖЈЁЃЦфжаВњЮя ![]() НазіСЌЫФСђЫсИљРызгЃЌЦфНсЙЙЪНШчЯТЃК
НазіСЌЫФСђЫсИљРызгЃЌЦфНсЙЙЪНШчЯТЃК
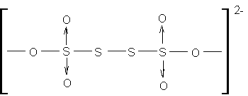
Ёя ХфЮЛзїгУЃК
![]()
СђДњСђЫсФЦДѓСПгУзїееЯрЕФЖЈгАМСЁЃееЯрЕзЦЌЩЯЮДИаЙтЕФфхЛЏвјдкЖЈгАвКжааЮГЩЁВAg(S2O3)2ЁГ3-ЖјШмНтЁЃ
ЂЧЙ§СђЫсМАЦфбЮ
Й§СђЫсПЩвдПДзїЪЧЙ§бѕЛЏЧтЕФбмЩњЮяЁЃЙ§бѕЛЏЧтНсЙЙЪНЮЊHЉЄOЉЄOЉЄH,ЛЧЫсЛљЉЄSO3HНсЙЙЪНЮЊЃК 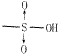 ,H2O2ЗжзгжаЕФСНИіЧтдзгБЛЉЄSO3HЗжВНШЁДњЃЌЗжБ№ЕУЕНЕФВњЮяЮЊЙ§вЛСђЫсH2SO5КЭЙ§ЖўСђЫсH2S2O8ЁЃ
,H2O2ЗжзгжаЕФСНИіЧтдзгБЛЉЄSO3HЗжВНШЁДњЃЌЗжБ№ЕУЕНЕФВњЮяЮЊЙ§вЛСђЫсH2SO5КЭЙ§ЖўСђЫсH2S2O8ЁЃ
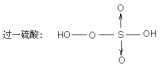
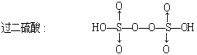
Й§СђЫсОпгаЧПбѕЛЏад(РДздЙ§бѕМќЉЄOЉЄOЉЄ)ЁЃЫќУЧзїЮЊбѕЛЏМСВЮгыЗДгІЕФЙ§ГЬжаЃЌЙ§бѕМќЖЯСбЃЌетСНИібѕдзгЕФбѕЛЏжЕгЩдРДЕФЃ1БфЮЊЃ2ЃЌЖјСђЕФбѕЛЏжЕБЃГжЃЋ6ВЛБфЁЃ
живЊЕФЙ§ЖўСђЫсбЮгаK2S2O8КЭ(NH4)2S2O8,ЫќУЧвВЪЧЧПбѕЛЏМСЁЃ
Й§СђЫсбЮШШЮШЖЈадВюЃЌР§ШчЃК
![]()
ЂШСЌЖўбЧСђЫсМАЦфбЮ
СЌЖўбЧСђЫсH2S2O4ЪЧЖўдЊЫсЃЌКмВЛЮШЖЈЃЌгіЫЎЛсСЂМДЗжНтЮЊСђКЭбЧСђЫсЁЃ
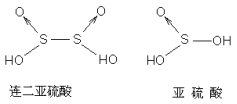
ЁЁ 2H2S2O4 + H2O Ёњ H2S2O3 + 2H2SO3
2H2S2O3 Ёњ S + H2SO3
СЌЖўбЧСђЫсФЦNa2S2O3ЁЄ2H2OЫзГЦБЃЯеЗлЃЌЦфЮШЖЈадБШЯргІЕФЫсЧПЁЃ
![]()
СЌЖўбЧСђЫсФЦЪЧЧПЛЙдМСЁЃNa2S2O4ФмНЋI2ЁЂCu2+ЁЂAg+ЕШЛЙдЃЌФмАбЯѕЛљЛЏКЯЮяЛЙдЮЊАБЛљЛЏКЯЮяЁЃПеЦјжаЕФбѕФмНЋNa2S2O3бѕЛЏЁЃ
2Na2S2O4 + O2 + 2H2OЁњ 2NaHSO3 + 2NaHSO4
СЌЖўбЧСђЫсФЦЪЧШОСЯЙЄвЕКЭгЁШОЙЄвЕЩЯГЃгУЕФЛЙдМСЃЌЛЙЙуЗКгУгкдьжНЁЂЪГЦЗЙЄвЕМАвНбЇЩЯЁЃ
ЂЗТШЛЧЫсКЭЖўТШЛЏСђѕЃ
ТШЛЧЫсHSO3ClПЩвдПДзїЮЊСђЫсЗжзгжаЕФвЛИієЧЛљБЛТШдзгШЁДњЕФВњЮяЁЃЦфНсЙЙЪНЮЊ:
ИЩдяЕФТШЛЏЧтЦјЬхгыЗЂбЬСђЫсЗДгІЩњГЩТШЛЧЫсЃК
HCl + SO3 Ёњ HSO3Cl
ТШЛЧЫсЪЧвЛжжЮоЩЋЁЂгаДЬМЄадЦјЮЖЕФЦјЬхЃЌгіЫЎЗЂЩњОчСвЫЎНтВЂБЌеЈЃЌдкГБЪЊЕФПеЦјжавђЫЎНтЖјЗЂбЬЁЃдкгаЛњЛЏбЇЩЯТШЛЧЫсЪЧСМКУЕФЛЧЛЏМСЁЃ
ЖўТШЛЏСђѕЃSO2Cl2ЯрЕБгкСђЫсЗжзгжаЕФСНИієЧЛљЖМБЛТШдзгШЁДњЕФВњЮяЃЌЦфНсЙЙЪНШчЯТЃК
ЖўбѕЛЏСђКЭТШЦјдкДпЛЏМСДцдкЬѕМўЯТФмжБНгЛЏКЯЩњГЩЖўТШЛЏСђѕЃЃК
![]()
ЖўТШЛЏСђѕЃвВЪЧвЛжжЮоЩЋвКЬхЃЌгаЧПСвЕФЫЎНтзїгУЁЃЖўТШЛЏСђѕЃвВгУгкгаЛњКЯГЩЙЄвЕЁЃ
ЕкЪЎЖўеТ ЕЊзхЁЂЬМзхКЭХ№зхдЊЫи
БОеТЮвУЧжївЊСЫНтЕЊзхЁЂЬМзхКЭХ№зхдЊЫиЕФЭЈадЃЌЪьЯЄЯрЙиЗжзгЕФНсЙЙКЭЮШЖЈадЁЃ
Ёь12.1 ЕЊзхдЊЫи
12.1.1 ЕЊзхдЊЫиИХЪі
ЕЊзхвВНаЂѕAзхЁЃ
ЕЊзхдЊЫиЗћКХЃК | N | P | As | Sb | Bi |
МлВуЕчзгЙЙаЭЃК | 2s22p3 | 3s23p3 | 4s24p3 | 5s25p3 | 6s26p3 |
бѕЛЏжЕЃК | -3ЁЋ+5 | -3,+3,+5 | -3,+3,+5 | (-3),+3,+5 | +3,(+5) |
зюДѓХфЮЛЪ§ЃК | 4 | 6 | 6 | 6 | 6 |
ЕЊзхдЊЫиЕФ МлВуЕчзгЙЙаЭЮЊns2np3ЃЌЕч ИКадВЛЪЧКмДѓЃЌЫљвдБОзхдЊЫиаЮГЩбѕЛЏжЕЮЊе§ЕФЛЏКЯЮяЕФЧїЪЦБШНЯУїЯдЃЌЛЏКЯЮяжЛвЊЪЧЙВМлаЭЕФЃЌЖјЧвдзггњаЁЃЌаЮГЩЙВМлМќЧїЪЦвВгњДѓЁЃ
ЕЊзхдЊЫидкздШЛНчЕФДцдкаЮЪНЃК
ЕЊЃК | жївЊвдЕЅжЪДцдкгкДѓЦјжаЁЃ |
СзЃК | жївЊвдСзЫсбЮаЮЪНЗжВМдкЕиПЧжаЃЌШчЃЌСзЫсИЦCa3(PO4)2ЃЌЕЊСзЛвЪЏ3Ca3(PO4)2ЁЄCaF2ЁЃ |
ЩщЁЂЬрЁЂющЃК | жївЊвдСђЛЏЮяПѓДцдкЃЌШч,алЛЦ4S4As4ЁЂЛдЬрПѓSb2S3ЁЂЛдющПѓBi2S3ЕШЁЃ |
12.1.2 ЕЊзхдЊЫиЕФЕЅжЪ
ЕЊЦјЪЧЮоЩЋЁЂЮоГєЁЂЮоЮЖЕФЦјЬхЁЃЮЂШмгкЫЎЃЌЗаЕуЮЊ-195.8ЁцЁЃГЃЮТЯТЕЊЦјЕФаджЪМЋВЛЛюЦУЃЌМгШШЪБЕЊЦјгыЛюЦУН№ЪєLiЁЂCaЁЂMgЕШЗДгІЃЌЩњГЩРызгаЭЛЏКЯЮяЁЃ
ЕЊЗжзгЪЧЫЋдзгЗжзгЃЌСНИіЕЊдзгвдШ§МќНсКЯЃЌЕчзгХХВМЮЊЃК ![]() ЃЌгЩгкNЁдNМќЕФМќФм(946kJ/mol)ЗЧГЃДѓЃЌЫљвдN2ЪЧзюЮШЖЈЕФЫЋдзгЗжзгЃЌЕЊЦјБэЯжГіИпЕФЛЏбЇЖшадЃЌвђДЫЕЊЦјГЃБЛгУзїБЃЛЄЦјЬхЁЃ
ЃЌгЩгкNЁдNМќЕФМќФм(946kJ/mol)ЗЧГЃДѓЃЌЫљвдN2ЪЧзюЮШЖЈЕФЫЋдзгЗжзгЃЌЕЊЦјБэЯжГіИпЕФЛЏбЇЖшадЃЌвђДЫЕЊЦјГЃБЛгУзїБЃЛЄЦјЬхЁЃ
СзЕФГЃМћЕФЭЌЫивьаЮЬхгаЃКАзСзЁЂКьСзКЭКкСзЁЃ
АзСзЪЧЭИУїЕФЁЂШсШэЕФРЏзДЙЬЬхЃЌгЩP4ЗжзгЭЈЙ§ЗжзгМфСІЖбЛ§Ц№РДЃЌУПИіСздзгЭЈЙ§ЦфpxЃЌpyКЭpzЙьЕРЗжБ№КЭСэЭт3ИіСздзгаЮГЩ3ИіІвМќЃЌМќНЧЁЯPPPЮЊ60ЁуЃЌЗжзгФкВПОпгаеХСІЃЌЦфНсЙЙВЛЮШЖЈЁЃЫљвдP4ЛЏбЇаджЪКмЛюЦУЃЌдкПеЦјжаздШМЃЌФмШмгкЗЧМЋадШмМСЁЃ
НЋАзСзИєОјПеЦјМгШШЕН400ЁцЪБПЩЕУЕНКьСзЁЃКьСзЕФНсЙЙНЯИДдгЁЃвЛжжЙлЕуШЯЮЊЃКP4ЗжзгжаЕФвЛИіPЁЊPМќЖЯСбКѓЯрЛЅСЌНгЦ№РДаЮГЩГЄСДНсЙЙЃЌЫљвдКьСзНЯЮШЖЈЃЌ400ЁцвдЩЯВХШМЩеЁЃКьСзВЛШмгкгаЛњШмМСЁЃ
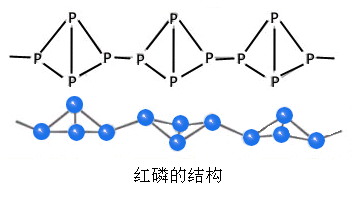
КкСзОпгагыЪЏФЋРрЫЦЕФВузДНсЙЙЃЌЕБгыЪЏФЋВЛЭЌЕФЪЧЃЌКкСзУПвЛВуФкЕФСздзгВЂВЛЖМдкЭЌвЛЦНУцЩЯЃЌЖјЪЧЯрЛЅСЌНгГЩЭјзДНсЙЙЁЃЫљвдКкСзОпгаЕМЕчадЃЌвВВЛШмгкгаЛњШмМСЁЃ

12.1.3 ЕЊЕФЛЏКЯЮя
ЂБ ЕЊЕФЧтЛЏЮя
дкЕЊЕФЧтЛЏЮяжажиЕубЇЯААБКЭяЇбЮЁЃ
ЂХАБ
НсЙЙЃКАБдзгвдШ§ИіВЛЕШаддгЛЏЙьЕРгыЧтдзгГЩМќЃЌаЮГЩШ§НЧзЖаЭНсЙЙЁЃ
жЦБИЃК
ЙЄвЕжЦБИЗЈЃК
![]()
ЪЕбщЪвжЦБИЗЈЃК
![]()
аджЪЃКАБЪЧЮоЩЋЦјЬхЃЌгаЬиЪтДЬМЄадЦјЮЖЃЌШмгкЫЎГЪМюадЁЃАБЕФЛЏбЇаджЪЛюЦУЃЌФмгыаэЖрЮяжЪЗЂЩњЗДгІЁЃ
бѕЛЏЛЙдЗДгІЃК
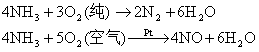
жївЊЬхЯжСЫАБЕФЧПЛЙдадЁЃ
МгКЯЗДгІЃК Ag++ 2NH3 Ёњ [Ag(NH3)2]+
ШЁДњЗДгІЃК ![]()
ЂЦяЇбЮ
яЇбЮвЛАуЮЊЮоЩЋОЇЬхЃЌОјДѓЖрЪ§взШмгкЫЎЃЌдкЫЎжаЖМгавЛЖЈГЬЖШЕФЫЎНтЃЌЫЎНтЗДгІЮЊЃК
![]()
![]() ЕФНсЙЙЃК
ЕФНсЙЙЃК ![]() жаЕФЕЊдзгВЩгУЫФИіsp3дгЛЏЙьЕРгыЫФИіЧтдзгГЩМќЃЌаЮГЩе§ЫФУцЬхЕФПеМфЙЙаЭЁЃ
жаЕФЕЊдзгВЩгУЫФИіsp3дгЛЏЙьЕРгыЫФИіЧтдзгГЩМќЃЌаЮГЩе§ЫФУцЬхЕФПеМфЙЙаЭЁЃ
![]() ЕФМјЖЈЃКЪЏШяЪджНЗЈКЭNesslerЪдМСЗЈЁЃдкЪдвКжаМгМюЃЌМгШШЪдвКЪЙАБЦјвнГіЃЌгУЪЏШяЪджНМьбщЃЌЪджНгЩКьБфРЖЃЌгУNesslerЪджНМьбщЃЌЪджНгЩАзЕНКьзидйЕНЩюКжЩЋЁЃЗДгІПЩБэЪОШчЯТЃК
ЕФМјЖЈЃКЪЏШяЪджНЗЈКЭNesslerЪдМСЗЈЁЃдкЪдвКжаМгМюЃЌМгШШЪдвКЪЙАБЦјвнГіЃЌгУЪЏШяЪджНМьбщЃЌЪджНгЩКьБфРЖЃЌгУNesslerЪджНМьбщЃЌЪджНгЩАзЕНКьзидйЕНЩюКжЩЋЁЃЗДгІПЩБэЪОШчЯТЃК
![]()
ЙЬЬхяЇбЮЕФШШЮШЖЈадВюЃКяЇбЮЕФШШЗжНтЧщПівђзщГЩяЇбЮЕФЫсЕФаджЪВЛЭЌЖјвьЁЃ
![]()
![]()
ЗЧЛгЗЂадЗЧбѕЛЏадЫсЕФяЇбЮ,ЗжНтЪБжЛгаАБЛгЗЂЕє,ЖјЫсЛђЫсЪНбЮдђСєдкШнЦїжаЁЃ
![]()
![]()
бѕЛЏадЫсЕФяЇбЮЃЌЗжНтГіЕФАБЦјБЛбѕЛЏГЩN2КЭN2OЁЃ
![]()
![]()
ЂВ ЕЊЕФбѕЛЏЮя
ЕЊЕФбѕЛЏЮяЗжзгжавђЫљКЌЕФNЃOМќНЯШѕ ЃЌЦфШШЮШЖЈадЖМБШНЯВюЃЌЫќУЧЪмШШвзЗжНтЛђвзБЛбѕЛЏЁЃЦфжазюживЊЕФЪЧNOЛђNO2ЁЃ
ЂХвЛбѕЛЏЕЊ(NO)
НсЙЙЃКNOЕФЗжзгЙьЕРЕчзгХХВМЮЊЃК(1Ів)2(2Ів)2(3Ів)2(4Ів)2(1Іа)4(2Іа)1ПЩМћЗжзгжагаЮДГЩЖдЕФЕЅЕчзгДцдкЃЌNOОпгаЫГДХадЁЃNOВЮгыЗДгІЪБШнвзЪЇШЅ2ІаЙьЕРЩЯЕФвЛИіЕЅЕчзгаЮГЩбЧЯѕѕЃРызгNO+ЁЃ
жЦБИЃКNOЪЧЯѕЫсЩњВњЕФжаМфВњЮяЁЃ
ЙЄвЕжЦЗЈЃК ![]()
ЪЕбщЪвжЦЗЈЃК 3Cu + 8HNO3(ЯЁ) Ёњ 3Cu(NO3)2 + 2NO + 4H2O
аджЪЃКNOЪЧЮоЩЋЦјЬхЃЌЫЎжаШмНтЖШНЯаЁЁЃ
2NO + O2 Ёњ 2NO2
2NO + Cl2 Ёњ 2NOCl(ТШЛЏбЧЯѕѕЃ)
Fe2+ + NO Ёњ [Fe(NO)]2+(бЧЯѕѕЃКЯЬњ(Ђё))
ЂЦЖўбѕЛЏЕЊКЭЫФбѕЛЏЕЊ
НсЙЙЃКNO2жаЕФNвдСНИіsp2дгЛЏЙьЕРгыбѕдзгГЩМќЃЌаЮГЩVзжаЭЕФПеМфЙЙаЭЃЌДЫЭтЗжзгжаЛЙаЮГЩвЛИіШ§жааФШ§ЕчзгДѓЁЧ( ![]() )МќЁЃ
)МќЁЃ
![]()
аджЪЃКNO2ЪЧгаЬиЪтГєЮЖЕФКьзиЩЋгаЖОЦјЬхЁЃ
NO2РфШДЪБзЊЛЏЮЊЮоЩЋЕФN2O4(g)ЁЃ
![]()
NO2БЛЫЎЮќЪеЩњГЩЯѕЫсКЭNOЃЌБЛNaOHЮќЪеЩњГЩЯѕЫсбЮКЭбЧЯѕЫсбЮЁЃ
3NO2 + H2O Ёњ 2HNO3 + NO
2NO2 +2NaOH Ёњ NaNO3 + NaNO2 + H2O
ЂГ ЕЊЕФКЌбѕЫсМАЦфбЮ
ЂХбЧЯѕЫсМАЦфбЮ
бЧЯѕЫсЕФНсЙЙЃКHNO2жаNвдСНИіsp2дгЛЏЙьЕРЗжБ№гыєЧЛљ
бѕКЭбѕдзгаЮГЩІвМќЃЌNКЭOдзгжЎМфЛЙаЮГЩвЛИіІаМќЃЌНсЙЙШчЭМЫљЪОЁЃ
бЧЯѕЫсЕФжЦБИЃКБиаыдкРфШмвКжажЦБИЁЃ
![]()
![]()
бЧЯѕЫсЕФаджЪЃКбЧЯѕЫсЪЧвЛжжШѕЫсЃЌ ![]() ЁЃбЧЯѕЫсМЋВЛЮШЖЈЃЌжЛФмДцдкгкКмЯЁЕФРфШмвКжаЃЌШмвКХЈЫѕЛђМгШШЪБЖМЛсЗжНтЁЃ
ЁЃбЧЯѕЫсМЋВЛЮШЖЈЃЌжЛФмДцдкгкКмЯЁЕФРфШмвКжаЃЌШмвКХЈЫѕЛђМгШШЪБЖМЛсЗжНтЁЃ
![]()
бЧЯѕЫсИљЕФНсЙЙЃК ![]() жаЕФNВЩШЁСНИіsp2дгЛЏЙьЕРЗжБ№гыбѕдзгаЮГЩІвМќЃЌДЫЭтЛЙаЮГЩвЛИіШ§жааФЫФЕчзгДѓЁЧМќ(
жаЕФNВЩШЁСНИіsp2дгЛЏЙьЕРЗжБ№гыбѕдзгаЮГЩІвМќЃЌДЫЭтЛЙаЮГЩвЛИіШ§жааФЫФЕчзгДѓЁЧМќ( ![]() ),РызгЪЧVзжаЭНсЙЙЁЃ
),РызгЪЧVзжаЭНсЙЙЁЃ
бЧЯѕЫсбЮЕФжЦБИЃКгУМюЮќЪеЕШЮяжЪЕФСПЕФNOКЭNO2ЁЃ
NO + NO2 + 2NaOH Ёњ 2NaNO2 + H2O
бЧЯѕЫсбЮЕФаджЪЃКГ§AgNO2ЪЧЧГЛЦЩЋВЛШмадЙЬЬхЭтЃЌДѓЖрЪ§бЧЯѕЫсбЮЪЧЮоЩЋЕФЃЌвЛАувзШмгкЫЎЃЌМЋЖОЃЌЪЧжТАЉЮяжЪЁЃ
бЧЯѕЫсбЮдкЫсадНщжЪжаМШгабѕЛЏадгжгаЛЙдадЃЌЪЕМЪгІгУжаГЃзїбѕЛЏМСЃЌЦфЛЙдВњЮявЛАуЮЊNOЁЃ
![]()
ЕБбЧЯѕЫсбЮгыЧПбѕЛЏМСзїгУЪБЃЌВХБэЯжГіЦфЛЙдадЁЃ
![]()
аЮГЩбЧЯѕЫсбЮЕФбєРызгН№ЪєаддНЧПЃЌбЧЯѕЫсбЮЕФЮШЖЈаддНИпЁЃР§ШчЃКAgNO2ЃМNaNO2ЁЃ
ЂЦЯѕЫсМАЦфбЮ
ЯѕЫсЕФНсЙЙЃКHNO3жаЕФNдзгВЩШЁsp2дгЛЏЙьЕРЗжБ№гывЛИієЧЛљбѕКЭСНИібѕдзгаЮГЩ3ИіІвМќЃЌNдзгжаЮДВЮгыдгЛЏЕФpЙьЕРжаЕФСНИіЕчзггыСНИіЗЧєЧЛљбѕдкOЃNЃOМфаЮГЩШ§жааФЫФЕчзгЁЧМќ( ![]() ),ШчЭМЫљЪОЁЃ
),ШчЭМЫљЪОЁЃ
ЯѕЫсЕФаджЪЃКХЈЯѕЫсКмВЛЮШЖЈЃЌМћЙтЪмШШвзЗжНтЁЃ
4HNO3 Ёњ 4NO2 + O2 +2H2O
ЯѕЫсгЩгкЛгЗЂЖјВњЩњАзбЬЃЌЙЪГЦЮЊЗЂбЬЯѕЫсЃЛЯѕЫсжаГЃвђШмгаЗжНтГіРДЕФNO2ЖјДјгаЛЦЩЋЛђКьзиЩЋЁЃ
ЯѕЫсзюЕфаЭЕФаджЪЪЧЫќЕФЧПбѕЛЏадЁЃЯѕЫсПЩвдАбЬМЁЂСзЁЂСђЁЂЕтЕШаэЖрЗЧН№ЪєбѕЛЏЮЊИпМлЫсЃЌЖјЦфздЩэБЛЛЙдЮЊNOЁЃ
3C + 4HNO3 Ёњ 3CO2 + 4NO + 2H2O
3P + 5HNO3 + 2H2 Ёњ 3H3PO4 + 5NO
S + 2HNO3 Ёњ H2SO4 + 2NO
3I2 + 10HNO3 Ёњ 6HIO3 + 10NO + 2H2O
ДѓВПЗжН№ЪєПЩШмгкЯѕЫсЃЌЯѕЫсБЛЛЙдЕФГЬЖШгыН№ЪєЕФЛюЦУадКЭЯѕЫсЕФХЈЖШгаЙиЁЃ
Cu + 4HNO3(ХЈ) Ёњ Cu(NO3)2 + 2NO2 + 2H2O
3Cu + 8HNO3(ЯЁ) Ёњ 3Cu(NO3)2 + 2NO + 4H2O
4Zn + 10NO3(ЯЁ) Ёњ 4Zn(NO3)2 + N2O + 5H2O
4Zn + 10NO3(КмЯЁ) Ёњ 4Zn(NO3)2 + NH4NO3 + 3H2O
ПЩМћЃЌН№ЪєдНЛюЦУЃЌЯѕЫсЕФХЈЖШдНЕЭЃЌHNO3БЛЛЙдКѓЕЊЕФбѕЛЏжЕдНЕЭЁЃ
Н№ЁЂВЌЕШВЛЛюЦУН№ЪєФмШмгкЭѕЫЎ(ЯѕЫсгыХЈбЮЫсвдЬхЛ§БШ1:3ЕФБШР§ЖјХфжЦЕФЛьКЯЮя)жаЁЃ
Au + HNO3 + 4HCl Ёњ H[AuCl4] + NO + 2H2O
3Pt + 4HNO3 + 18HCl Ёњ 3H2[PtCl6] + 4NO +8H2
зЂвтЃКРфЕФХЈЯѕЫсПЩвдЪЙFeЁЂAlЁЂCrЖлЛЏ(ХЈЯѕЫсНЋН№ЪєБэУцбѕЛЏГЩвЛВуБЁЖјжТУмЕФбѕЛЏЮяБЃЛЄФЄЃЌжТЪЙН№ЪєВЛФмдйгыЯѕЫсМЬајзїгУ)ЁЃ
ХЈЯѕЫсЛЙФмгыгаЛњЛЏКЯЮяЗЂЩњЯѕЛЏЗДгІЁЃ
![]()
ЯѕЫсИљЕФНсЙЙЃК ![]() жаЕФNВЩШЁsp2дгЛЏЙьЕРЗжБ№гы3ИіOдзгаЮГЩІвМќЃЌNжаЕФЮЊВЮгыдгЛЏЕФpЙьЕРжаЕФСНИіЕчзггы3ИіOЕФ3ИіpЕчзгаЮГЩЫФжааФСљЕчзгДѓЁЧМќ(
жаЕФNВЩШЁsp2дгЛЏЙьЕРЗжБ№гы3ИіOдзгаЮГЩІвМќЃЌNжаЕФЮЊВЮгыдгЛЏЕФpЙьЕРжаЕФСНИіЕчзггы3ИіOЕФ3ИіpЕчзгаЮГЩЫФжааФСљЕчзгДѓЁЧМќ( ![]() )ЁЃ
)ЁЃ
ЯѕЫсбЮЕФаджЪЃКМИКѕЫљгаЕФЯѕЫсбЮЖМШмгкЫЎЁЃЯѕЫсбЮЙЬЬхЛђЫЎШмвКдкГЃЮТЯТЖМБШНЯЮШЖЈЃЌЫЎШмвКдкЫсадЬѕМўЯТВХгабѕЛЏадЃЌЙЬЬхдкИпЮТЪБВХгабѕЛЏадЃЌЙЪЯѕЫсбЮПЩгУзїеЈвЉЁЂЛ№вЉЁЂбцЛ№жаЕФбѕЛЏМСЁЃ
ЯѕЫсбЮЪмШШЪБЗжНтЃЌЗжНтВњЮявђН№ЪєРызгаджЪВЛЭЌЖјЗжЮЊШ§РрЃК(вдН№ЪєЛюЖЏЫГађжаЕФХХађЮЊзМ)
KЁЋMgЃК ![]()
MgЁЋCuЃК ![]()
CuвдКѓЃК ![]()
ЫМПМЃКБШНЯЯѕЫсбЧЯѕЫсЕФЫсадЁЂбѕЛЏадЃЛБШНЯЯѕЫсбЮЁЂбЧЯѕЫсбЮЕФШШЮШЖЈадЁЃ
12.1.4 СзЕФЛЏКЯЮя
ЂБ СзЕФЛЏКЯЮя(ьЂPH3)
НсЙЙЃКШ§НЧзЖаЮЃЌРрЫЦгкNH3ЁЃ
жЦБИЃКЕЅжЪСздкМюШмвКжаЦчЛЏЁЃ
P4 + 3KOH + 3H2O Ёњ PH3 + 3KH2PO2(ДЮСзЫсМи)
ДЫЭтЛЙПЩгУРрЫЦжЦБИАБЕФЗНЗЈжЦБИPH3ЁЃ
ИДЗжНтЗЈЃКPH4I + KOH Ёњ PH3 + H2O + KI
ЫЎНтЗЈЃК Mg3P2 + 6H2O Ёњ 2PH3 + 3Mg(OH)2
аджЪЃКьЂЪЧЮоЩЋЦјЬхЃЌгаЫЦДѓДаГєЮЖЃЌОчЖОЁЃгаЧПЛЙдадЁЃ
PH3 + 2O2 Ёњ H3PO4
ЂВСзЕФбѕЛЏЮя
СзЕФГЃМћбѕЛЏЮягаЮхбѕЛЏЖўСзКЭШ§бѕЛЏЖўСзЁЃ
ЂХШ§бѕЛЏЖўСз
СздкбѕЦјВЛзуЕФЬѕМўЯТШМЩеЩњГЩP4O6,МђГЦЮЊШ§бѕЛЏЖўСзЁЃШ§бѕЛЏЖўСзЕФНсЙЙШчЭМЫљЪОЁЃЯрЕБгк6ИібѕдзгЁАВхЁБдкP4ЕФЫФУцЬхЕФСљИіРтЩЯЁЃЫљвдP4O6ЪЧАзЩЋвзЛгЗЂЕФРЏзДЙЬЬхЃЌвзШмгкгаЛњШмМСЁЃ
P4O6ЪЧбЧСзЫсєћЃЌгыРфЫЎЗДгІНЯТ§ЃЌЩњГЩбЧСзЫсЁЃ
P4O6 + 6H2O(Рф) Ёњ 4H3PO3
P4O6дкШШЫЎжаЦчЛЏЮЊСзЫсКЭьЂ(ЛђЕЅжЪСз)ЁЃ
P4O6 + 6H2O(ШШ) Ёњ 3H3PO4 + PH3
5P4O6 + 182O(ШШ) Ёњ 12H3PO4 + 8P
P4O6гкбѕЦјНјвЛВНЗДгІЩњГЩP4O10ЁЃ
P4O6 + 2O2 Ёњ P4O10
ЂЦЮхбѕЛЏЖўСз
СздкГфзуЕФПеЦјжаШМЩеЩњГЩP4O10,МђГЦЮЊЮхбѕЛЏЖўСзЁЃЮхбѕЛЏЖўСзЕФНсЙЙШчЭМЫљЪОЁЃЯрЕБгкдкP4O6ЕФЛљБОНсЙЙЕЅдЊЕФИїИіСздзгЕФЖЅЩЯдйМгЩЯвЛИібѕдзгЃЌP4O10ЪЧбЉЛЈзДОЇЬхЃЌЧвгаКмЧПЕФЮќЫЎадЃЌГЃгУзїИЩдяМСЁЃ
P4O10 + 6H2O Ёњ 4H3PO4
P4O10 + 6H2SO4 Ёњ 6SO3 + 4H3PO4
P4O10 + 12HNO3 Ёњ 6N2O5 + 4H3PO4
ПЩМћP4O10ПЩЪЙСђЫсЁЂЯѕЫсЕШЭбЫЎЩњГЩбѕЛЏЮяЁЃ
ЂГ СзЕФКЌбѕЫсМАЦфбЮ
СзФмаЮГЩЖржжКЌбѕЫсЁЃСзЕФКЌбѕЫсАДбѕЛЏжЕВЛЭЌПЩЗжЮЊДЮСзЫсH3PO2ЁЂбЧСзЫсH3PO3КЭСзЫсH3PO4ЕШЁЃ
ЂХДЮСзЫсМАЦфбЮ
аджЪЃКДЮСзЫсЪЧвЛдЊжаЧПЫсЃЌ ![]() ЁЃH3PO2ЪЧЧПЛЙдМСЃЌФмдкШмвКжаНЋAgNO3ЁЂHgCl2ЁЂCuCl2ЕШжиН№ЪєбЮЛЙдЮЊН№ЪєЕЅжЪЁЃ ДЮСзЫсбЮЖрвзШмгкЫЎЃЌвВЪЧЧПЛЙдМСЁЃ
ЁЃH3PO2ЪЧЧПЛЙдМСЃЌФмдкШмвКжаНЋAgNO3ЁЂHgCl2ЁЂCuCl2ЕШжиН№ЪєбЮЛЙдЮЊН№ЪєЕЅжЪЁЃ ДЮСзЫсбЮЖрвзШмгкЫЎЃЌвВЪЧЧПЛЙдМСЁЃ
НсЙЙЃК
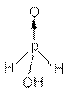
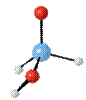
ЂЦбЧСзЫсМАЦфбЮ
аджЪЃКH3PO3ЪЧЖўдЊжаЧПЫсЃЌ ![]() ЃЌ
ЃЌ ![]() ЁЃ
ЁЃ
H3PO3ЪЧЧПЛЙдМСЃЌЪмШШвзЦчЛЏЁЃ
H3PO3 + 2Ag+ + H2O Ёњ H3PO4 + 2Ag + 2H+
![]()
НсЙЙЃК
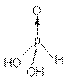
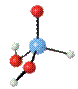
бЧСзЫсФмаЮГЩе§бЮКЭЫсЪНбЮЁЃМюН№ЪєКЭИЦЕФбЧСзЫсбЮвзШмгкЫЎЃЌЦфЫћН№ЪєЕФбЧСзЫсбЮЖМФбШмЁЃбЧСзЫсбЮвВЪЧНЯЧПЕФЛЙдМСЁЃ
ЂЧСзЫс
аджЪЃКH3PO4ЪЧШ§дЊжаЧПЫсЃЌ ![]() ЃЌ
ЃЌ ![]() ЃЌ
ЃЌ ![]() ЁЃ
ЁЃ
е§СзЫсПЩаЮГЩШ§жжРраЭЕФбЮЃКе§бЮЁЂСзЫсвЛЧтбЮЁЂСзЫсЖўЧтбЮЁЃвЛАуе§СзЫсбЮБШНЯЮШЖЈЃЌВЛвзЗжНтЁЃ
НсЙЙЃК
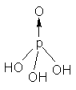
СзЫсЕФШ§жжбЮРрШмНтадКЭЫЎНтадБШНЯШчЯТЃК
| M3PO4 | M2HPO4 | MH2PO4 |
ШмНтадЃК | ДѓЖрЪ§ФбШмгкЫЎ(Г§K+,Na+,яЇРызгЭт) | ДѓЖрЪ§взШмгкЫЎ | |
ЫЎШмвКЫсМюадЃК | PHЃО7 | PHЃО7 | PHЃМ7 |
двђЃК | ЫЎНтЮЊжї | ЫЎНтЃОНтРы | ЫЎНтЃМНтРы |
СзЫсбЮжазюживЊЕФЕФЪЧИЦбЮЁЃЙЄвЕЩЯРћгУЬьШЛСзЫсИЦЩњВњСзЗЪЃЌЗДгІШчЯТЃК
Ca3(PO4)2 + 2H2SO4 + 4H2O Ёњ Ca(H2PO4)2 + 2(CaSO4ЁЄ2H2O)(Й§СзЫсИЦЃЈСзЗЪЃЉ)
![]() ЕФМјЖЈЃКНЋСзЫсбЮгыЙ§СПЕФютЫсяЇ(NH4)2MoO4МАЪЪСПЕФХЈЯѕЫсЛьКЯКѓМгШШЃЌПЩТ§Т§ЩњГЩЛЦЩЋЕФСзютЫсяЇГСЕэЃЌЗДгІЮЊЃК
ЕФМјЖЈЃКНЋСзЫсбЮгыЙ§СПЕФютЫсяЇ(NH4)2MoO4МАЪЪСПЕФХЈЯѕЫсЛьКЯКѓМгШШЃЌПЩТ§Т§ЩњГЩЛЦЩЋЕФСзютЫсяЇГСЕэЃЌЗДгІЮЊЃК
![]()
СзЫсЪмШШКѓЭбЫЎПЩаЮГЩНЙСзЫсЁЂОлСзЫсЁЂЦЋСзЫсЕШЁЃ
НЙСзЫсМАЦфбЮ
СНИіСзЫсЗжзгЭбШЅвЛИіЫЎЗжзгМАЕУНЙСзЫсH4P2O4ЁЃНЙСзЫсЪЧЮоЩЋВЃСЇзДЮяжЪЃЌвзШмгкЫЎЁЃдкРфЫЎжаЛКТ§зЊЛЏЮЊСзЫсЁЃ
H4P2O4 + H2O Ёњ 2H3PO4
НЙСзЫсЮЊЫФдЊжаЧПЫс ЃЌНсЙЙЮЊЃК 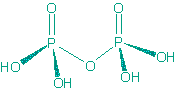
ГЃМћЕФНЙСзЫсбЮЮЊM2H2P2O7КЭM4P2O7СНРрЁЃ ![]() ОпгаХфЮЛФмСІЃЌР§ШчЃК
ОпгаХфЮЛФмСІЃЌР§ШчЃК
![]()
![]()
ЫљвдНЙСзЫсбЮПЩгУгкгВЫЎШэЛЏКЭЮоЧшЕчЖЦЁЃ
ЦЋСзЫсМАЦфбЮ
ЦЋСзЫсжИвЛИіH3PO4ЭбШЅвЛИіH2OМДHPO3ЁЃШєЮЊnИіH3PO4ЭбШЅnИіH2OЃЌМДЮЊЖрОлЦЋСзЫсЃЌГЃМћЕФгаШ§ОлЦЋСзЫсКЭЫФОлЦЋСзЫсЁЃЭбЫЎЙ§ГЬвдЫФОлЦЋСзЫс(HPO3)4ЮЊР§ЃЌЭМЪОШчЯТЃК
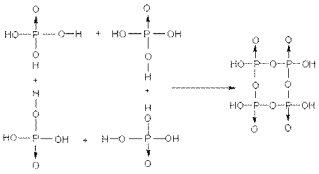
ОлСзЫсЪЧжИnИіH3PO4ЭбШЅnЃ1ИіЫЎЃЌШчНЙСзЫсМДЮЊЖўОлСзЫсЃЛШ§ОлСзЫсЕФаЮГЩПЩЭМЪОШчЯТЃК
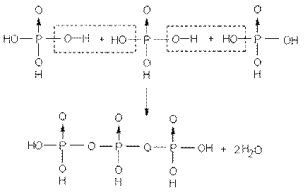
ОлСзЫсбЮЭЌбљОпгаХфЮЛФмСІЁЃ
![]()
аЁНсЃК
Ёя ЫѕКЯЖШдіМгЃЌЫсаддіЧПЁЃ
| H5P3O10 | H4P2O7 | H3PO4 |
|
| 2.9ЁС10-2 | 6.7ЁС10-3 |
| 10-2 | 5.3ЁС10-3 | 6.2ЁС10-8 |
| 10-3 | 2.2ЁС10-7 | 4.5ЁС10-13 |
Ёя МјБ№е§СзЫсЁЂНЙСзЫсКЭЦЋСзЫсЕФЗНЗЈЃК
| е§СзЫс | НЙСзЫс | ЦЋСзЫс |
AgNO3 | ЛЦЩЋЁ§ | АзЩЋЁ§ | АзЩЋЁ§ |
ЕААзжЪ |
|
| АзЩЋЁ§ |
ЂДСзЕФТБЛЏЮя
СзПЩвдаЮГЩШ§ТБЛЏСзPX3КЭЮхТБЛЏСзPX5ЁЃ
зюживЊЕФТБЛЏЮяЮЊШ§ТШЛЏСзКЭЮхТШЛЏСзЁЃдкЪвЮТЯТЃЌШ§ТШЛЏСзЪЧЮоЩЋвКЬхЃЌЮхТШЛЏСзЪЧАзЩЋОЇЬхЃЌИУОЇЬхЪЧЮоЩЋОЇЬхЃЌОЇЬхжаКЌга[PCl4]+КЭ[PCl6]-РызгЁЃ
СзЕФТБЛЏЮязюживЊЕФаджЪОЭЪЧЫЎНтадЁЃ
PCl3 + 3H2O Ёњ H3PO3 +3HCl
PCl5 + 4H2O Ёњ H3PO4 +5HCl
| PCl3 | PCl5 |
НсЙЙ | Ш§НЧзЖ | Ш§НЧЫЋзЖ |
|
|
|
жааФдзгдгЛЏРраЭ | ВЛЕШадsp3 | sp3d |
12.1.5 ЩщЁЂЬрЁЂющЕФЛЏКЯЮя
ЂБЩщЁЂЬрЁЂющЕФЧтЛЏЮя
ЩщЁЂЬрЁЂющЕФЧтЛЏЮяЗжБ№ЪЧЃКыЯAsH3ЁЂ ![]() SbH3ЁЂ
SbH3ЁЂ ![]() BiH3ЁЃетаЉЧтЛЏЮяЖМЪЧЮоЩЋвКЬхЃЌНсЙЙгыАБРрЫЦЃЌОљЮЊШ§НЧзЖаЮЁЃаджЪБфЛЏЙцТЩШчЯТЃК
BiH3ЁЃетаЉЧтЛЏЮяЖМЪЧЮоЩЋвКЬхЃЌНсЙЙгыАБРрЫЦЃЌОљЮЊШ§НЧзЖаЮЁЃаджЪБфЛЏЙцТЩШчЯТЃК
Ёя ШлЁЂЗаЕуДгыЯЕН ![]() гЩЕЭЕНИпЁЃ
гЩЕЭЕНИпЁЃ
Ёя ЮШЖЈадДгыЯЕН ![]() гЩИпЕНЕЭЁЃ
гЩИпЕНЕЭЁЃ
Ёя МюадДгыЯЕН ![]() гЩЧПЕНШѕЁЃ
гЩЧПЕНШѕЁЃ
ыЯЁЂ ![]() ЁЂ
ЁЂ ![]() ОљМЋЖОЁЃыЯгаЫЦДѓЫтЕФДЬМЄадЦјЮЖЃЌЪвЮТЯТдкПеЦјжаШМЩеЃК
ОљМЋЖОЁЃыЯгаЫЦДѓЫтЕФДЬМЄадЦјЮЖЃЌЪвЮТЯТдкПеЦјжаШМЩеЃК
2AsH3 + 3O2 Ёњ As2O3 +3H2
дкШБбѕЬѕМўЯТЃЌыЯЪмШШЗжНтЃК
![]()
МьбщЩњЮяЬхЪЧЗёКЌЩщжаЖОЃЌРћгУЕФЪЧAsH3ЕФШШВЛЮШЖЈадКЭЛЙдадЁЃ
ЂВ ЩщЁЂЬрЁЂющЕФбѕЛЏЮя
ЩщЁЂЬрЁЂющПЩаЮГЩСНРрбѕЛЏЮяЃЌМДM2O3КЭM2O5ЃЌM2O3ЪЧЯргІЕФбЧЫсЕФЫсєћЃЌM2O5ЪЧЯргІЕФе§ЫсЕФЫсєћЁЃ
ЩщЁЂЬрЁЂющЕФЕЅжЪдкПеЦјжаШМЩеКЭБКЩеЫќУЧЕФСђЛЏЮяПЩжЦЕУЫќУЧЕФM2O3ЁЃ
| As2O3 | Sb2O3 | Bi2O3 |
беЩЋЁЂзДЬЌ | АзЩЋЗлФЉ | АзЩЋЙЬЬх | ЛЦЩЋЗлФЉ |
ЫЎШмад | ЮЂШм | ФбШм | МЋФбШм |
ЫсМюад | СНадЦЋЫс | СНад | Мюад |
ОЇЬхНсЙЙ | ЗжзгОЇЬх | ЗжзгОЇЬх | РызгОЇЬх |
ГЃЮТЮЊЫЋОлЗжзг | As4O6 | Sb4O6 |
|
Ш§бѕЛЏЖўЩщAs2O3ЃЌЫзГЦХјЫЊЃЌОчЖОЁЃжївЊгУгкжЦдьЩБГцМСЁЂГ§ВнМСвдМАКЌЩщвЉЮяЁЃ
As2O5ЮЊАзЩЋЃЛSb2O5ЮЊЕЛЦЩЋЃЛBi2O5ЮЊКьзиЩЋЃЌМЋВЛЮШЖЈЁЃ
ЂГЩщЁЂЬрЁЂющЕФЧтбѕЛЏЮяМАЦфКЌбѕЫс
ЫсМюадЕнБфЃК

M(Ђѓ)ЕФЛЙдадЃК
M(Ђѕ)ЕФбѕЛЏадЃК
зЂвтЯТУцЗДгІЗЂЩњЕФЬѕМўЃЌВЂНтЪЭЮЊЪВУДЃП
![]()
![]()
ЂДЩщЁЂЬрЁЂющЕФбЮ
ЩщЁЂЬрЁЂющФбвдаЮГЩM5+ЃЌЕЋдкЧПЫсШмвКжаПЩвдаЮГЩM3+ЁЃЩщЁЂЬрЁЂющЕФбЮдкЫЎжаЖМвзЫЎНтЁЃAsCl3ЕФЫЎНтРрЫЦгкPCl3,ВњЮяЮЊСНжжЫсЁЃ
AsCl3 + 3H2O Ёњ H3AsO3 + 3HCl
ЬрЁЂющЕФбЮЫЎНтЮЊМюЪНбЮЁЃ
Sb2(SO4)3 + 2H2OЁњ (SbO)2SO4(s) + 2H2SO4
BiCl3 + H2O Ёњ BiOCl(s) + 2HCl
Sb3+ЁЂBi3+ОпгавЛЖЈЕФбѕЛЏадЁЃ
2Sb3+ + 2Sn Ёњ 2Sb + 3Sn2+
етЪЧМјЖЈSb3+ЕФЬиеїЗДгІЁЃ
2Bi3+ + 3[Sn(OH)4]2- + 6OH- Ёњ 2Bi + 3[Sn(OH)6]2-
етЪЧМјЖЈBi3+ЕФЬиеїЗДгІЁЃ
ЂЕЩщЁЂЬрЁЂющЕФСђЛЏЮя
СђЛЏЮяЃК
As2S3(ЛЦ) | Sb2S3(ГШ) | Bi2S3(Кк) |
As2S5(ЛЦ) | Sb2S5(ГШ) | Bi2S5(ВЛФмаЮГЩ) |
СђЛЏЮяЕФаджЪЃК
ЂХВЛШмгкЫЎКЭЯЁЫсЁЃ
ЂЦХфЮЛШмНт(ХЈHCl)ЁЃ
![]()
![]()
![]()
As2S3ЁЂAs2S5ВЛШмЁЃ
ЂЧМюШм
NaOH
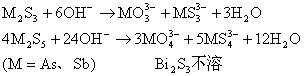
Na2S
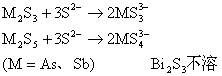
ЂШбѕЛЏМюШм
![]()
ЂЩгыбѕЛЏадЫс(HNO3)зїгУ
![]()
Ёь12.2ЁЁЬМзхдЊЫи
12.2.1 ЬМзхдЊЫиИХЪі
ЬМзхдЊЫиЗћКХЃК | C | Si | Ge | Sn | Pb |
МлВуЕчзгЙЙаЭЃК | 2s22p2 | 3s23p2 | 4s24p2 | 5s25p2 | 6s26p2 |
бѕЛЏжЕ | -4,+2,+4 | +4 | (+2),+4 | +2,+4 | +2,(+4) |
зюДѓХфЮЛЪ§ЃК | 4 | 4 | 4 | 6 | 6 |
ЬМзхдЊЫиЕФМлВуЕчзгЙЙаЭЮЊ2s22p2ЃЌЕУЪЇЕчзгЖМНЯФбЃЌетОіЖЈСЫЬМзхдЊЫиГЩМќЕФжївЊЧуЯђЪЧЙВМлМќЁЃ
ЬМзхдЊЫидкздШЛНчЕФДцдкаЮЪНЃК
ЬМЃКН№ИеЪЏЁЂЪЏФЋЃЛУКЁЂЪЏгЭЁЂЬьШЛЦјЃЛЬМЫсбЮЃЛCO2
ЙшЃКSiO2КЭЙшЫсбЮЁЃ
ерЃКжївЊвдСђЛЏЮяаЮЪНДцдкЃК4Ag2SЁЄGeS2(СђвјерПѓ)КЭ2PbSЁЄGeS2(СђЧІерПѓ),ЧызЂвтЧІКЭерЕФбѕЛЏжЕЃЌ
ЬхЛсЕНЖшадЕчзгЖдаЇгІЕФДцдкЁЃ
Ю§ЃКЮ§ЪЏ(SnO2)
ЧІЃКЗНЧІПѓ(PbS)ЃЌАзЧІПѓ(PbCO3)
12.2.2 ЬМзхдЊЫиЕФЕЅжЪ
ЕЅжЪЬМДцдкзХЭЌЫивьаЮЬхЃК
ЮоЖЈаЭЬМЃК ФОЬПЁЂНЙЬПЁЂЙЧЬМЁЃ
Н№ИеЪЏЃК дзгОЇЬхЃЌгВЖШДѓЃЌШлЕуИпЁЃ
ЪЏФЋЃК ВузДОЇЬхЁЃ
зуЧђЯЉ(ИЛРеЯЉ)ЃК C60ЃЌC70ЕШЁЃ
ЕЅжЪЙшвВДцдкзХЭЌЫивьаЭЬхЃКЮоЖЈаЭЬхКЭОЇЬхЙшЃЌКѓепЪЧдзгОЇЬхЁЃ
ЕЅжЪерЃКЪЧЛвАзЩЋН№ЪєЃЌНсЙЙРрЫЦгкН№ИеЪЏЁЃ
ЕЅжЪЮ§ЃКШ§жжЭЌЫивьаЭЬхЃК
![]()
ЕЭЮТЯТЃЌАзЮ§БфГЩЗлФЉЃЌГЦжЎЮЊ"Ю§вп"ЁЃ
ЧІЃКжЪШэЃЌФмзшЕВXЩфЯпЃЌПЩвдгУзїКЫЗДгІЖбЕФБЃЛЄЦСЁЃ
12.2.3 ЬМЕФЛЏКЯЮя
1. ЬМЕФбѕЛЏЮя
ЂХвЛбѕЛЏЬМ(CO)
НсЙЙЃКCOгыN2ЮЊЕШЕчзгЬх(14e-),НсЙЙЯрЫЦЃЌОљгаШ§МќЁЃ
![]()
![]()
аджЪЃК
Ђй зїЮЊХфЮЛЬхаЮГЩєЪЛљХфКЯЮяЃЌFe(CO)5ЃЌNi(CO)4ЃЌCo2(CO)8ЃЌЦфжаCЪЧХфЮЛдзгЁЃ
Ђк COзїЮЊЛЙдМСЃЌдквБН№ЙЄвЕжаЕУЕНЙуЗКгІгУЁЃ
3CO(g) + Fe2O Ёњ 2Fe(s) + 3CO2(g)
CO(g) + CuO(s) Ёњ Cu(s) + CO2(g)
CO(g) + PdCl2(aq) + H2O Ёњ Pd(s) + CO2 + 2HCl(aq)
ДЫЗДгІвђЩњГЩКкЩЋPbЃЌПЩгУгкМьбщCOЕФДцдкЁЃ
Ђл COгаЖОЃЌПеЦјжаCOЕФЬхЛ§БШДяЕН1ЈM800ЃЌШЫдкАыаЁЪБФЧОЭЛсЫРЭіЁЃвђЮЊCOгыбЊвКжаЕФбЊКьЕААзНсКЯЃЌЪЙжЎЪЇШЅдибѕФмСІЃЌШЫШБбѕЖјжЯЯЂЫРЭіЁЃ
ЂЦЖўбѕЛЏЬМ(CO2)
ОЕфЕФЗжзгНсЙЙЃКO=C=OЃЌКѓРДШЫУЧВтЕУCO2жаCгыOМфЕФМќГЄЮЊ116pmЃЌНщгкЫЋМќМќГЄгыШ§МќМќГЄжЎМф( ![]() жаЕФC=OМќГЄЮЊ124pmЃЌCOШ§МќМќГЄЮЊ113pm)ЃЌЮЊДЫШЫУЧЬсГіCO2жагІДцдквдЯТМќаЭЃКCгыOМфГ§СЫгаІвМќЭтЃЌдкCO2
жаЕФC=OМќГЄЮЊ124pmЃЌCOШ§МќМќГЄЮЊ113pm)ЃЌЮЊДЫШЫУЧЬсГіCO2жагІДцдквдЯТМќаЭЃКCгыOМфГ§СЫгаІвМќЭтЃЌдкCO2
ЗжзгжаЩаДцдк2ИіШ§жааФЫФЕчзгДѓЁЧМќ( ![]() )
) ![]()
2. ЬМЫсМАЦфбЮ
CO2ШмгкЫЎЃЌДѓВПЗжвдCO2·�������H2OаЮЪНДцдкЃЌМЋаЁВПЗжЪЧвдH2CO3ЕФаЮЪНДцдкЃЌвВОЭЪЧЫЕCO2ШмгкЫЎКѓЃЌЫЎШмвКжаДцдквдЯТЦНКтЃК
![]()
![]()
![]()
гІжИГіЕФЪЧЃЌЩЯЪіНтРыГЃЪ§ЪЧАбCO2ПДзїШЋВПаЮГЩЬМЫсЃЌетгыЪТЪЕВЛЗћЃЌПМТЧШмвКжаЕФЦНКтЃЌH2CO3ЕФЪЕМЪНтРыГЃЪ§ЮЊЃК ![]() ЁЃ
ЁЃ
Ёя ЬМЫсбЮЕФШШЮШЖЈадЃК
Ђй е§бЮ>ЫсЪНбЮ>ЫсЃЌМДM2CO3ЃОMHCO3ЃОH2CO3ЁЃР§ШчNa2CO3КмФбЗжНтЃЌNaHCO3дк270ЁцЗжНтЃЌH2CO3дкЪвЮТЯТМДПЩЗжНтЁЃ
![]()
![]()
H2CO3 Ёњ H2O + CO2
Ђк ЭЌвЛзхЕФН№ЪєЕФЬМЫсбЮЕФЮШЖЈадДгЩЯЕНЯТдіМгЁЃ
Р§ШчЃК
ЬМЫсбЮ | BeCO3 | MgCO3 | CaCO3 | SrCO3 | BaCO3 |
ЗжНтЮТЖШ/Ёц | 100 | 540 | 900 | 1290 | 1360 |
r(M2+)діДѓЃЌЮШЖЈаддіМгЃЌЗжНтЮТЖШдіИпЁЃ
Ђл Й§ЖЩН№ЪєЬМЫсбЮЮШЖЈадВю
ЬМЫсбЮ | CaCO3 | PbCO3 | ZnCO3 | FeCO3 |
ЗжНтЮТЖШ/Ёц | 900 | 315 | 350 | 282 |
M2+МлВуЕчзгЙЙаЭ | 8e- | (8+2)e- | 18e- | (9-17)e- |
ЩЯЪіШШЮШЖЈадЙцТЩПЩгУРызгМЋЛЏРэТлНтЪЭЃК
дк ![]() ФкВПЃКC(4+)ЖдO2-гаМЋЛЏФмСІЃЌЖјдкM2+гы
ФкВПЃКC(4+)ЖдO2-гаМЋЛЏФмСІЃЌЖјдкM2+гы ![]() жЎМфЃК
жЎМфЃК 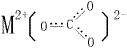
M2+ЖдСкНќЕФO2-гаЗДМЋЛЏзїгУЃЌЕжЯћC(4+)ЖдO2-ЕФМЋЛЏзїгУЃЌЯћШѕСЫCЁЄЁЄЁЄOМќЃЌЗДМЋЛЏзїгУЕФДѓаЁгыM2+ЕФАыОЖЃЌМлВуЕчзгЙЙаЭгаЙиЃЌM2+ЕФАыОЖдНаЁ(ШчBe2+)ЃЌМЋЛЏСІдНЧПЃЌMOНсКЯдНУмЧаЃЌCЃOМќЖЯПЊЃЌЗжНтЮТЖШОЭдНЕЭЃЛЗЧ8e-ЙЙаЭM2+МЋЛЏСІДѓЃЌЫљвдЃЌЗжНтЮТЖШвВЕЭЁЃ
Ёя ЬМЫсбЮЕФШмНтадЃК
ЖдФбШмбЮРДЫЕЃЌЫсЪНбЮШмНтЖШДѓгке§бЮЃЌР§ШчЃКCa(HCO3)2взШмгкЫЎЃЌЖјCaCO3ФбШмгкЫЎЁЃЕиБэВужаЕФЬМЫсбЮПѓЪЏдкCO2КЭЫЎЕФГЄЦкЧжЪДЯТФмВПЗжЕФзЊБфЮЊCa(HCO3)2ЖјШмНтЁЃ
CaCO3+ CO2 + H2O Ёњ Ca(HCO3)2
ЪЏЛвбвЕигђГіЯжжгШщЪЏОАЙлгыДЫгаЙиЁЃ
ЖдвзШмбЮРДЫЕЃЌЙцТЩЧЁКУЯрЗДЃЌЫсЪНбЮШмНтЖШаЁгке§бЮЁЃ
ЬМЫсбЮ | Na2CO3 | NaHCO3 | K2CO3 | KHCO3 |
S (100Ёц)g/100g H2O | 45 | 16 | 156 | 60 |
етЪЧгЩгкЧтМќДцдкЃЌ ![]() жЎМфаЮГЩЖўОлЮяЛђЖрОлЮя дЕЙЪЁЃ
жЎМфаЮГЩЖўОлЮяЛђЖрОлЮя дЕЙЪЁЃ
Ёя ЬМЫсбЮЕФЫЎНтадЃК
![]()
![]()
ШмвКжаКЌга ![]() ЁЂOHЃЕШЃЌДЫЪБЃЌМгШыН№ЪєРызгЩњГЩЕФГСЕэРраЭЪЧЪВУДЃП
ЁЂOHЃЕШЃЌДЫЪБЃЌМгШыН№ЪєРызгЩњГЩЕФГСЕэРраЭЪЧЪВУДЃП
Н№ЪєРызгМгШыЬМЫсбЮКѓЃЌЩњГЩГСЕэЕФРраЭЃК
Ђй ЧтбѕЛЏЮяМюадЧПЕФЕФН№ЪєРызггыжЎЗДгІЩњГЩЬМЫсбЮГСЕэЁЃР§ШчЃКBa2+ЃЌSr2+ЃЌCa2+КЭAg+ЕШЁЃ
![]()
Ђк ЧтбѕЛЏЮяМюадНЯШѕЕФН№ЪєРызггыжЎЗДгІЩњГЩЬМЫсєЧбЮ(МюЪНЬМЫсбЮ)ГСЕэЁЃР§ШчЃЌPb2+ЃЌBi3+ЃЌCu2+ЃЌCd2+ЃЌZn2+ЃЌHg2+ЃЌCo2+ЃЌNi2+КЭMg2+ЕШЁЃ
![]()
![]()
Ђл ЫЎНтадЧПЃЌСНадЕФН№ЪєРызггыжЎЗДгІЩњГЩЧтбѕЛЏЮяГСЕэЁЃР§ШчЃКAl3+ЃЌFe3+ЃЌCr3+ЃЌSn2+ЃЌSn4+,Sb3+ЕШЁЃ
![]()
![]()
 12.2.4 ЙшЕФЛЏКЯЮя
12.2.4 ЙшЕФЛЏКЯЮя
1.ЙшЕФбѕЛЏЮяЃКSiO2(ЙшЪЏ)
ЮоЖЈаЭЬхЃКЪЏгЂВЃСЇЁЂЙшдхЭСЁЂьнЪЏЁЃ
ОЇЬхЃКЬьШЛSiO2ОЇЬхГЦЮЊЪЏгЂЃЌДПОЛЕФЪЏгЂГЦЮЊЫЎОЇЃЌЪєгкдзгОЇЬхЃЌгУгкжЦзїЙтбЇвЧЦїМАЙЄвеЦЗЕШЁЃ
КЌдгжЪЕФЪЏгЂЃКТъшЇЁЂзЯОЇЁЃ
SiO2ЕФаджЪЃКSiO2ЪєгкЫсадбѕЛЏЮяЃЌЪдЯыЫќгыМюЗДгІЕФВњЮяЁЃСэЭтЃЌSiO2гыHFЗДгІЕФЪЕгУадЃК
SiO2 + 2NaOH Ёњ Na2SiO3 + H2O
SiO2 + Na2CO3 Ёњ Na2SiO3 + CO2
SiO2 + 4HF Ёњ SiF4 + 2H2O
2.ЙшЫсМАЦфбЮ
ЙшЫсЃКH2SiO3(дЙшЫсH4SiO4ЕФЭбЫЎаЮЪН)ЖўдЊШѕЫсЃК ![]() ЃЌПЩМћЙшЫсЫсадБШЬМЫсШѕЃЌгЩДЫПЩЯыЕНжЦБИЙшЫсПЩгУЫсадБШЙшЫсЧПЕФЖўбѕЛЏЬМЁЂТШЛЏяЇЁЂТШЛЏЧтЕШгыЙшЫсФЦЗДгІЃК
ЃЌПЩМћЙшЫсЫсадБШЬМЫсШѕЃЌгЩДЫПЩЯыЕНжЦБИЙшЫсПЩгУЫсадБШЙшЫсЧПЕФЖўбѕЛЏЬМЁЂТШЛЏяЇЁЂТШЛЏЧтЕШгыЙшЫсФЦЗДгІЃК
Na2SiO3 + 2NH4Cl Ёњ H2SiO3 + 2NaCl + 2NH3
Na2SiO3 + 2HCl Ёњ H2SiO3 + 2NaCl
ВњЮяH2SiO3ЃЌПЊЪМЮЊЕЅЗжзгЙшЫсШмгкЫЎЃЌКѓОлКЯГЩЖрЙшЫсЃЌаЮГЩЙшЫсШмНКЃЌЕБХЈЖШДѓЪБЛђМгШыЕчНтжЪЪБЃЌаЮГЩФ§НКЃЌОЙ§ИЩдяКѓЕУЕНАзЩЋЭИУїЙЬЬхЁЃЫќОпгааэЖрМЋЯИаЁЕФПзЯЖЃЌБэУцЛ§ДѓЃЌЮќИНФмСІЧПЃЌГЃгУзїИЩдяМСКЭЮќИНМСдиЬхЁЃНўЭИЙ§CoCl2ЕФЙшНКГЦЮЊБфЩЋЙшНКЃЌИЩдяЪБГЪРЖЩЋЃЌЮќЫЎБЅКЭКѓГЪЗлКьЩЋЁЃ
ЙшЫсбЮЃКГ§СЫNa2SiO3(ЫзГЦЫЎВЃСЇ)ЮЊПЩШмадЙшЫсбЮЭтЃЌДѓЖрЪ§ЙшЫсбЮФбШмгкЫЎЃЌЧвгаЬиеїбеЩЋЁЃ ЫЎжаЛЈдАЃЈФбШмгкЫЎЕФЙшЫсбЮЃЉ
ЙшЫсбЮНсЙЙИДдгЃЌвдSiO4ЫФУцЬхЮЊНсЙЙЕЅдЊЃЌПЩСЌНгГЩЯпВуЁЂжїЬхЭјзДЁЃ вЛАувдбѕЛЏЮяаЮЪНБэЪОЙшЫсбЮЃЌ
Р§ШчЃКАздЦЪЏ K2OЁЄ3Al2O3ЁЄ6SiO2ЁЄ2H2O
ХнЗаЪЏ Na2OЁЄAl2O3ЁЄ2SiO2ЁЄnH2O
ТСЙшЫсбЮ ШЫЙЄКЯГЩЕФТСЙшЫсбЮГЦЮЊЗжзгЩИЃЌЬьШЛХнЗаЪЏ гыШЫЙЄКЯГЩЗжзгЩИЖМОпгаЖрПзЖрбЈНсЙЙЃЌжБОЖБШПзбЈаЁЕФЗжзгФмНјШыПзбЈжаЃЌжБОЖБШПзЕРДѓЕФЗжзгБЛОмжЎгкЭтЃЌЦ№зХЩИбЁЗжзгЕФзїгУЁЃШЫУЧПЩИљОнВЛЭЌашвЊжЦБИВЛЭЌНсЙЙЁЂВЛЭЌПзОЖЕФЗжзгЩИЁЃЙуЗКгУгкЪЏгЭЛЏЙЄЁЂвБН№вНСЦЕШЙЄвЕЁЃ
3.ЙшЕФТБЛЏЮя
ТБЛЏЮя | SiF4 | SiCl4 | SiBr4 | SiI4 |
ОлМЏзДЬЌ | g | l | l | s |
ЦфЗаЕувРДЮЩ§ИпЃЌЪдгУбЇЙ§ЕФжЊЪЖНтЪЭИУЙцТЩЁЃ
ЫЎНтадЃК SiCl4 + 3H2O Ёњ H2SiO3 + 4HCl
SiF4 + 3H2O Ёњ H2SiO4 + 4HF
SiF4 + 2HF Ёњ H2[SiF6]
12.2.5 Ю§ЁЂЧІЕФЛЏКЯЮя
1.бѕЛЏЮяЕФЫсМюад
ОљЮЊСНад,ЕБЫсМюЧПЖШВЛЕШЁЃ
M(Ђђ)ЃК ![]()
![]()
![]() (МгHNO3ЛђHAcВЛФмМгHClЁЂH2SO4)
(МгHNO3ЛђHAcВЛФмМгHClЁЂH2SO4)
![]()
M(Ђє)ЃК
ІСЃЮ§ЫсФмШмгкЫсвВФмШмгкМюЃЌІТЃЮ§ЫсВЛШмгкЫсвВВЛШмгкМюЁЃ
Sn + 4HNO3(ХЈ) Ёњ H2SnO3 + 4NO2 + H2O
змжЎЃЌЮ§ЁЂЧІЕФЧтбѕЛЏЮяЕФЫсМюадЙцТЩШчЯТЃК
Ёя ЭЌвЛдЊЫиИпбѕЛЏжЕЧтбѕЛЏЮяБШЕЭбѕЛЏжЕЧтбѕЛЏЮяЕФЫсадЧПЃЛ
Ёя ЭЌвЛбѕЛЏжЕЕФВЛЭЌдЊЫиЃЌзёбЭЌвЛжїзхгЩЩЯЕНЯТМюаддіЧПЕФЙцТЩЁЃЯрЖдЖјбдЃЌPb(OH)2МюадзюЧПЃЌSn(OH)4ЫсадзюЧПЃЌЫљвдГЦжЎЮЊЮ§ЫсЁЃ

2.Ю§ЁЂЧІ(ЂђЁЂЂє)ЛЏКЯЮяЕФбѕЛЏЛЙдад
![]()
![]()
Sn(Ђђ)ОпгаЧПЛЙдад
ЫсадЃК
ДЫЗДгІЪЧМјЖЈSn2+ЛђHg2+ЕФЗДгІЁЃ
МюадЃК
![]()
ДЫЗДгІЪЧМјЖЈBi3+ЕФЗДгІЁЃ
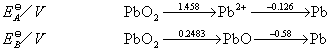
Pb(Ђє)ОпгаЧПбѕЛЏад
5PbO2 + 2Mn2+ + 4H+ Ёњ 2MnO4Ѓ + 5Pb2+ + 2H2O
3PbO2 Ёњ Pb3O4 + O2
Pb3O4ЃЌЫзГЦЧІЕЄЃЌКьЩЋЁЃгыбЧТщШЪгЭЛьКЯКѓзїЮЊгЭ ЛвЭПдкЙмзгЕФЯЮНгДІЃЌЗРжЙТЉЫЎЁЃPb3O4ПЩПДзїЪЧPb2PbO4(дЧІЫсЕФPb(Ђђ)бЮ)
Pb2PbO4+ 4HNO3 Ёњ 2Pb(NO3)2 + PbO2 + H2O
3.Ю§ЁЂЧІЕФбЮРр
ЂХЫЎНт
Ю§ЁЂЧІЕФЧтбѕЛЏЮяЪЧСНадЕФЃЌвђДЫЦфбЮРрвзЫЎНтЃК
Sn2+ + H2O + 2Cl- Ёњ Sn(OH)Cl(s,Аз) + HCl
Sn4+ + 6H2O Ёњ H2[Sn(OH)6]( ІСЃЮ§Ыс) + 4H+
![]()
ЂЦЧІ(Ђђ)бЮЕФШмНтад
ЩйЪ§ПЩШмЃК | Pb(NO3)2,Pb(Ac)2(ЮЖЬ№ЃЌЫзГЦЧІЬЧШѕЕчНтжЪ)ЃЌПЩШмадЧІбЮОљгаЖОЁЃ |
ЖрЪ§ФбШмЃК | PbCl2ЃЌPbSO4ЃЌPbI2(Н№ЛЦ)ЃЌPbCrO4(ЛЦ)ЁЃ |
ШмНтЗНЗЈЃК | PbCl2ШмгкШШЫЎКЭХЈHCl(ЩњГЩ |
| PbSO4ШмгкХЈH2SO4ЃКPbSO4 + H2SO4 Ёњ Pb(HSO4)2 |
| PbI2ШмгкKIЃКPbI2 + 2KI Ёњ K2PbI4 |
| PbCrO4ШмгкHNO3ЃК |
| врШмгк МюЃК |
4.ЧІЕФСђЛЏЮя
SnS(зи)ЃЌSnS2(ЛЦ)ЃЌPbS(Кк)ЃЌВЛДцдкPbS2ЁЃ
ЂХОљВЛШмгкЫЎЃЌВЛШмгкЯЁHClЁЃ
ЂЦШмгкХЈHCl(ХфЮЛШмНт)
![]()
![]()
![]()
ЂЧМюШм(SnSЃЌPbSМюадЧПЃЌВЛШмгкМю)
![]()
![]()
ЂШбѕЛЏМюШм(SnS2ЃЌPbSВЛШм)
![]()
ЂЩбѕЛЏШмНт
![]()
Ёь12.3ЁЁХ№зхдЊЫи
12.3.1 Х№зхдЊЫиИХЪі
Х№зх(ЂѓA)АќРЈЃКХ№ЃЈBЃЉ,ТСЃЈAlЃЉ, яиЃЈGaЃЉ, юїЃЈInЃЉ, юшЃЈTlЃЉЃЌМлЕчзгЙЙаЭЮЊЃКns2np1 ЃЌХ№зхдЊЫиЕФМлЕчзгЪ§ЃЈ3ИіЕчзгЃЉ<МлВуЙьЕРЪ§ЃЈ1ИіsЙьЕРИі+3ИіpЙьЕРЃЉЃЌЫљвдХ№зхдЊЫиЪЧШБЕчзгдЊЫиЁЃЫљвдХ№зхдЊЫивзаЮГЩШБЕчзгЛЏКЯЮяЁЃ Р§ШчЃКBF3
ШБЕчзгЛЏКЯЮяЃК ГЩМќЕчзгЖдЪ§<МлВуЙьЕРЪ§
ШБЕчзгЛЏКЯЮяЪЧЕфаЭЕФLewisЫсЃЌ
a. взаЮГЩХфЮЛЛЏКЯЮяЃЌР§ШчЃКHF+BF3ЁњHBF4
 b. взаЮГЩЫЋОлЮяЃЌР§ШчЃКAl2Cl6
b. взаЮГЩЫЋОлЮяЃЌР§ШчЃКAl2Cl6
12.3.2 Х№зхдЊЫиЕФЕЅжЪ
ЕЅжЪХ№гаЖржжЭЌЫивьаЮЬх,ЮоЖЈаЮХ№ЮЊзиЩЋЗлФЉЃЌОЇЬхХ№ГЪЛвКкЩЋЃЎЕЅжЪХ№ЕФгВЖШНќЫЦгкН№ИеЪЏЃЌХ№ЕФЕчзшКмДѓЃЌЕЋЫќЕФЕчЕМТЪШДЫцзХЮТЖШЕФЩ§ИпЖјдіДѓЁЃ
ОЇЬЌЕЅжЪХ№гаЖржжБфЬхЃЌЫќУЧЖМвдB12е§ЖўЪЎУцЬхЮЊЛљБОЕФНсЙЙЕЅдЊЁЃИУЖўЪЎУцЬхгЩ12ИіBдзгзщГЩЃЌ20ИіНгНќЕШБпШ§НЧаЮЕФРтУцЯрНЛГЩ30ЬѕРтБпКЭ12ИіНЧЖЅЃЌУПИіНЧЖЅЮЊвЛИіBдзгЫљеМОнЁЃгЩгкB12ЖўЪЎУцЬхЕФСЌНгЗНЪНВЛЭЌЃЌМќвВВЛЭЌЃЌаЮГЩЕФХ№ОЇЬхРраЭвВВЛЭЌЁЃ
ІСвЛСтаЮХ№ЪЧгЩB12ЕЅдЊзщГЩЕФВузДНсЙЙЃЌИУОЇЬхЪєгкдзгОЇЬхЃЌвђДЫОЇЬЌЕЅжЪХ№ЕФгВЖШДѓЃЌШлЕуИпЃЌЛЏбЇаджЪВЛЛюЦУЃЛЮоЖЈаЮХ№дђБШНЯЛюЦУЁЃ
ТСЪЧвјАзЩЋгаЙтдѓЕФЧсН№ЪєЃЌОпгаСМКУЕФЕМЕчадКЭбгеЙадЃЛЪЧКмживЊЕФН№ЪєВФСЯКЭНсЙЙВФСЯЃЌГЃгУвджЦзїЕМЯпЁЂКЭШегУЦїУѓЁЃ
яиЁЂюїЁЂюшЖМЪЧШэН№ЪєЃЌЮяРэаджЪЯрНќЃЌШлЕуЖМНЯЕЭЃЌяиЕФШлЕуБШШЫЬхЬхЮТЛЙЕЭЁЃяиЁЂюїЁЂюшПЩгУгкЩњВњаТаЭАыЕМЬхВФСЯЁЃ
12.3.3 Х№ЕФЛЏКЯЮя
ЯТУцСаОйСЫвЛаЉживЊХ№ЕФЛЏКЯЮя
Х№ЛЏЮя | беЩЋКЭзДЬЌ | УмЖШ(g/cm-3) | ШлЕу(Ёц) | ЪмШШЪБЕФБфЛЏ | ШмНтЖШЃЈg/100g H2O) |
ЫФХ№ЫсФЦ ЃЈХ№ЩАЃЉNaB4O7ЁС10H2O | МсгВАзЩЋОЇЬх | 1.73 ЃЈЮоЫЎЮЊ2.4ЃЉ |
| МгШШКѓЪзЯШШлЛЏЃЌШЛКѓХђеЭЪЇШЅНсОЇЫЎЁЃзюКѓБфЮЊВЃСЇзДЮяжЪЁЃNaB4O7ЁСH2Oдк200ЁцЪБШдЮШЖЈЃЌ400ЁцвдЩЯВХЭъШЋЭбЫЎЁЃ | 2.01ЃЈ20ЁцЃЉЫЎШмвКГЪМюадЃЌвВФмШмгкИЪгЭЃЌШДВЛШмгкОЦОЋЁЃ |
Х№Ыс H3BO3 | СљНЧаЮОЇЬхЃЌЗЂжщЙтЕФАзЩЋСлзДЮя | 1.46 | 171.0 | 107ЁцЪБЪЇШЅЫЎЃЌБфЮЊЦЋХ№ЫсHBO2ЃЌдк140Ёц~160ЁцЪББфЮЊНЙХ№ЫсH2B4O7ЃЈЫФХ№ЫсЃЉЃЌКьШШЪББфЮЊB2O3 | 4.74ЃЈ20ЁцЃЉ23.3ЃЈ90ЁцЃЉШмгкЫЎКѓЃЌгае§ЁЂЦЋКЭНЙаЭЕФРызгЃЌвВФмШмгкИЪгЭЃЌОЦОЋМАУбжа |
Ш§бѕЛЏЖўХ№ B2O3 | ЮоЩЋЭИУїВЃСЇзДЮя | 2.55 | 450.0 |
| 1.1ЃЈ0ЁцЃЉ15.7ЃЈ100ЁцЃЉМЋвзЮќЫЎЃЌЮќЪЊКѓБфЮЊЛьзЧзДЬЌЃЌвВФмШмгкОЦОЋ |
Ш§ЗњЛЏХ№ BF3 | ЮоЩЋЦјЬх | 3.077g/L(STP) | -127.1 | -100ЁцЗаЬк | дкЫЎжаШмНт |
ЕЊЛЏХ№ BN | АзЩЋЃЌЯёЛЌЪЏвЛбљЕФЗжзДЮя | 2.18 2.34 | 2967 3300ЃЈМгбЙЃЉ | 4930ЁцЩ§ЛЊМгШШЪБФбБЛбѕЛЏЁЃвВВЛгыCl2зїгУЃЌКьШШЪБгыH2OзїгУЗХГіNH3 | ВЛШмгкЫЎЃЌВЛгыЫсЃЌЧПМюШмвКзїгУЁЃ |
1. Х№ЕФЧтЛЏЮя
Х№гыЧтПЩаЮГЩаэЖрЙВМлаЭЧтЛЏЮяЃЌАДзщГЩПЩЗжЮЊСНДѓЯЕСаЃЌЭЈЪНЗжБ№ЮЊЃКBnHn+6(B4H10),BnHn+4(B2H6ЃЌB5H9ЃЌB6H10)ЃЌГЦЮЊХ№ЭщЁЃзюМђЕЅЕФХ№ЭщЪЧввХ№ЭщЃЈB2H6ЃЉЁЃХ№ЧтЛЏКЯЮяЕФЗжзгНсЙЙВЛФмНігУвЛАуЕФЙВМлМќРДБэЪОЁЃвђЮЊХ№дзгЪЧШБЕчзгдзгЃЌХ№ЭщЗжзгФкЫљгаМлЕчзгЕФЪ§ВЛФмТњзувЛАуЙВМлМќЫљашвЊЕФЪ§ФПЁЃдкB2H6ЗжзгжаЃЌЙВга14ИіМлЙьЕРЃЌЖјжЛга12ИіМлЕчзгЃЌЫљвдB2H6ЪЧШБЕчзгЛЏКЯЮяЁЃ
ввХ№ЭщЕФНсЙЙЃК дкB2H6ЗжзгжаЃЌга8ИіМлЕчзггУгк2ИіBдзгИїгы2ИіHдзгаЮГЩ4ИіBЉЄHІвМќЃЌет4ИіІвМќдкЭЌвЛЦНУцЩЯЁЃЪЃЯТЕФ4 ИіМлЕчзгдк2ИіBдзгКЭСэЭт2ИіHдзгжЎМфаЮГЩСЫДЙжБгкЩЯЪіЦНУцЕФ2ИіШ§жааФСНЕчзгМќЃЌвЛИідкЦНУцжЎЩЯЃЌСэвЛИідкЦНУцжЎЯТЃЌУПвЛИіШ§жааФСНЕчзгМќЪЧгЩ1ИіHдзгКЭ2ИіBдзгЙВгУ2ИіЕчзгЙЙГЩЕФЁЃетИіHдзгАб2ИіBдзгСЌНгЦ№РДЃЌОпгаЧХзДНсЙЙЃЌЮвУЧГЦетИіHдзгЮЊЁАЧХЧтдзгЁБЁЃ
ввХ№ЭщЕФаджЪЃК ГЃЮТЯТЃЌB2H6КЭB4H10ЃЈЖЁХ№ЭщЃЉЮЊЦјЬхЃЌB5~B8ЮЊвКЬхЃЌB10H14МАЦфЫќИпХ№ЭщЖМЪЧЙЬЬхЁЃХ№ЭщЖрЪ§гаЖОЃЌгаСюШЫВЛЪЪЕФЬиЪтЦјЮЖЃЌЧвВЛЮШЖЈЁЃввХ№Эщ(B2H6)ЪЧЮоЩЋЦјЬхЃЌгУLiHЃЌNaHЛђNaBH4гыТБЛЏХ№зїгУПЩвджЦБИB2H6
ЃЈ1ЃЉB2H6ЪЧЗЧГЃЛюЦУЕФЮяжЪЃЌБЉТЖгкПеЦјжавзШМЩеЛђБЌеЈЃЌВЂЗХГіДѓСПЕФШШЃК
![]()
ЃЈ2ЃЉ B2H6взЫЎНтЪЭЗХГіЧтЦјЃЌЩњГЩХ№ЫсЃК ![]()
ЃЈ3ЃЉB2H6гыLiHЗДгІ,ФмЩњГЩвЛжжБШB2H6ЕФЛЙдадИќЧПЕФЛЙдМСХ№ЧтЛЏяЎLiBH4 ЃК
![]()
LiBH4ЮЊАзЩЋбЮаЭЧтЛЏЮяЃЌШмгкЫЎЛђввДМЃЌЮоЖОЃЌЛЏбЇаджЪЮШЖЈЁЃЙуЗКгУгкгаЛњКЯГЩЃЌЪЧживЊЕФЛЙдМСЁЃ
ЃЈ4ЃЉЩњГЩХфКЯЮяЃК
![]()
2. Х№ЕФКЌбѕЛЏКЯЮя
гЩгкХ№гыбѕаЮГЩЕФB-OМќМќФмДѓЃЈB-OМќМќФмЮЊЃК523kJЁСmol-1ЃЉЃЌЫљвдХ№ЕФКЌбѕЛЏКЯЮяОпгаКмИпЕФЮШЖЈадЁЃЙЙГЩХ№ЕФКЌбѕЛЏКЯЮяЕФЛљБОЕЅдЊЪЧЦНУцШ§НЧаЮЕФBO3КЭЫФУцЬхаЮЕФBO4ЃЌетЪЧгЩХ№дЊЫиЕФЧзбѕадКЭШБЕчзгаджЪЫљОіЖЈЕФЁЃ
ЃЈ1ЃЉШ§бѕЛЏЖўХ№ЃЈB2O3ЃЉ
B2O3ЪЧАзЩЋЙЬЬхЃЌОЇЬЌB2O3БШНЯЮШЖЈЃЌШлЕуЮЊ460ЁцЁЃB2O3ФмБЛМюН№ЪєЁЂУОЁЂТСЛЙдЮЊЕЅжЪХ№ЃК
B2O3+ MgЁњ2B + 3MgO
B2O3взШмгкЫЎЃЌЩњГЩХ№ЫсЃК
B2O3+ 3H2O Ёњ 2H3BO3
ЕЋдкШШЕФЫЎеєЦјжадђЩњГЩЛгЗЂадЕФЦЋХ№ЫсHBO2ЃК
B2O3+ H2OЁњ 2HBO2
жЦБИB2O3ЕФвЛАуЗНЗЈЪЧМгШШХ№ЫсH3BO3ЪЙжЎЭбЫЎЃК
![]()
ЃЈ2ЃЉХ№Ыс
Х№ЫсАќРЈдХ№ЫсH3BO3ЁЂЦЋХ№ЫсH3BO2КЭЖрХ№ЫсxB2O3ЁСyH2OЁЃдХ№ЫсЭЈГЃгжГЦЮЊХ№ЫсЁЃНЋДПЕФХ№ЩА(Na2B4O7ЁЄ10H2O)ШмгкЗаЫЎжаВЂМгШыбЮЫсЃЌЗХжУКѓПЩЮіГіХ№ЫсЃК
Na2B4O7+ H2SO4 + 5H2OЁњ 4H3BO3+Na2SO4
НсЙЙЃКдкH3BO3ЕФОЇЬхжаЃЌУПИіBдзгвдШ§Иіsp2дгЛЏЙьЕРгыШ§ИіOдзгНсКЯГЩЦНУцШ§НЧаЮНсЙЙЃЌУПИіOдзгГ§вдЙВМлМќгы1ИіBдзгКЭ1ИіHдзгЯрНсКЯЭтЃЌЛЙЭЈЙ§ЧтМќгыСэвЛИіH3BO3ЕЅдЊжаЕФHдзгНсКЯЖјСЌГЩЦЌВуНсЙЙЃЌВугыВужЎМфдђвдЮЂШѕЕФЗЖЕТЛЊСІЯрЮќв§ЁЃЫљвдХ№ЫсОЇЬхЪЧЦЌзДЕФЃЌгаЛЌФхИаЃЌПЩзїШѓЛЌМСЁЃ
аджЪЃКH3BO3ЪЧАзЩЋЦЌзДОЇЬхЃЌЮЂШмгкЫЎЃЈ0ЁцЪБШмНтЖШЮЊ6.35g/(100gH2O)ЃЉЃЌЕЋдкШШЫЎжаШмНтЖШНЯДѓЃЈ100ЁцЪБШмНтЖШЮЊ27.6g/(100gH2O)ЃЉЁЃ
H3BO3ЪЧвЛдЊШѕЫсЃЌгЩгкBЪЧШБЕчзгдзгЃЌЫќМгКЯСЫРДздH2OЗжзгжаЕФOHЖјЪЭЗХГіРызгЃК
H3BO3+ H2O ![]() B(OH)4+ H+
B(OH)4+ H+ ![]() =5.8ЁС10-10
=5.8ЁС10-10
БэУїСЫХ№ЫсЪЧШБЕчзгЛЏКЯЮяЃЌЪЧвЛИіЕфаЭЕФТЗвзЪПЫсЁЃЦфЫсадПЩвђМгШыИЪТЖДМЛђИЪгЭЃЈБћШ§ДМЃЉЖјДѓЮЊдіЧПЃК
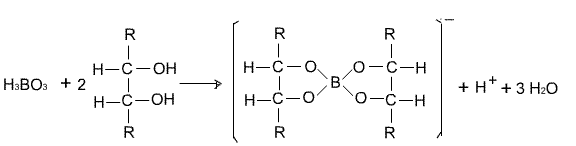
Х№ЫсКЭМзДМЛђввДМдкХЈH2SO4ДцдкЕФЬѕМўЯТЃЌЩњГЩХ№ЫсѕЅЃК
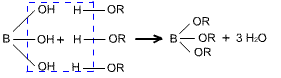
Х№ЫсѕЅдкИпЮТЯТШМЩеЛгЗЂЃЌВњЩњЬигаЕФТЬЩЋЛ№бцЃЌДЫЗДгІПЩгУгкМјЖЈКЌХ№ЛЏКЯЮяЕФДцдкЁЃ
Х№ЫсМгШШЭбЫЎЯШЩњГЩЦЋХ№ЫсHBO2ЃЌМЬајМгШШБфГЩB2O3ЁЃ
2H3BO3 ![]() 2HBO2+2H2O
2HBO2+2H2O ![]() B2O3+ 3H2O
B2O3+ 3H2O
ЃЈ3ЃЉХ№ЫсбЮ
Х№ЫсбЮгаЦЋХ№ЫсбЮЁЂдХ№ЫсбЮЁЂКЭЖрХ№ЫсбЮЕШЁЃзюживЊЕФХ№ЫсбЮЪЧЫФХ№ЫсФЦЃЌЫзГЦХ№ЩАЁЃХ№ЩАЕФЗжзгЪНЮЊNa2B4O5(OH)4ЁЄ8H2OЯАЙпЩЯвВГЃаДзїNa2B4O7ЁЄ10H2OЃЉЁЃ
ЫФХ№ЫсИљбєРызгЕФСЂЬхНсЙЙЃКдкЫФХ№ЫсИљжагаСНИіBO3ЦНУцШ§НЧаЮКЭСНИіBO4ЫФУцЬхЭЈЙ§ЙВгУНЧЖЅOдзгЖјСЊНсЦ№РДЁЃ
аджЪЃКХ№ЩАЪЧЮоЩЋАыЭИУїЕФОЇЬхЛђАзЩЋНсОЇЗлФЉЁЃдкПеЦјжаШнвзЪЇЫЎЗчЛЏЃЌМгШШЕН350Ёц~400ЁцзѓгвЃЌЪЇШЅШЋВПНсОЇЫЎГЩЮоЫЎбЮЃЌдк878ЁцШлЛЏЮЊВЃСЇЬхЁЃШлШкзДЬЌЕФХ№ЩАФмШмНтвЛаЉН№ЪєбѕЛЏЮяЃЌаЮГЩЦЋХ№ЫсбЮЃЌВЂвРН№ЪєЕФВЛЭЌЖјЯдЪОГіЬиеїбеЩЋЃЌР§ШчЃК
Na2B4O7 + CoO ![]() Co(BO2)2ЁЄ2NaBO2 ЃЈРЖЩЋЃЉ
Co(BO2)2ЁЄ2NaBO2 ЃЈРЖЩЋЃЉ
Na2B4O7 + NiO ![]() Ni(BO2)2ЁЄ2NaBO2 ЃЈзиЩЋЃЉ
Ni(BO2)2ЁЄ2NaBO2 ЃЈзиЩЋЃЉ
ДЫЗДгІПЩгУгкКИНгН№ЪєЪБГ§атЃЌвВПЩвдМјЖЈФГаЉН№ЪєРызгЃЌетдкЗжЮіЛЏбЇЩЯГЦЮЊХ№ЩАжщЪдбщЁЃ
Х№ЩАЪЧвЛИіЧПМюШѕЫсбЮЃЌПЩШмгкЫЎЃЌдкЫЎШмвКжаЫЎНтЖјЯдЪОНЯЧПЕФМюадЃК
[B4O5(OH)4]2- + 5H2O ![]() 4H3BO3+2OH-
4H3BO3+2OH- ![]() 2H3BO3 + 2B(OH)4-
2H3BO3 + 2B(OH)4-
Х№ЩАЫЎНтЪБЕУЕНЕШЮяжЪЕФСПЕФЫсЃЈH3BO3ЃЉКЭМюЃЈB(OH)4-ЃЉЃЌЫљвдДЫЫЎШмвКОпгаЛКГхзїгУЁЃХ№ЩАвзгкЬсДПЃЌЫЎШмвКгжЯдМюадЃЌдкЪЕбщЪвжаГЃгУЫќХфжЦЛКГхШмвКЛђзїЮЊБъЖЈЫсХЈЖШЕФЛљзМЮяжЪЁЃдкЙЄвЕЩЯЛЙПЩгУзіЗЪдэКЭЯДвТЗлЕФЬюСЯЁЃ
Х№ЩАжщЪдбщПЩПДзїЪЧШлШкЕФЫсадбѕЛЏЮяB2O3КЭМюадН№ЪєбѕЛЏЮязїгУЩњГЩгаЬиеїбеЩЋЕФЦЋХ№ЫсбЮВЃСЇЃЌЫљвдB2O3вВгаХ№ЩАжщЪдбщЃЌР§ШчЃК
B2O3 + CuO ![]() Cu(BO2)2 РЖЩЋ
Cu(BO2)2 РЖЩЋ
B2O3 + NiO ![]() Ni(BO2)2ТЬЩЋ
Ni(BO2)2ТЬЩЋ
дкBO3ЦНУцШ§НЧаЮНсЙЙжаЃЌBдзгвдsp2дгЛЏЙьЕРгыOдзгНсКЯЁЃЃЈШчЯТгвЭМЫљЪОЃЉ
дкBO4ЫФУцЬхНсЙЙжаЃЌBдзгвдsp3дгЛЏЙьЕРгыOдзгНсКЯЁЃЃЈШчЯТзѓЭМЫљЪОЃЉ
![]()
3 Х№ЕФТБЛЏЮя
Х№гыЧтПЩаЮГЩаэЖрЙВМлаЭЧтЛЏЮяЃЌАДзщГЩПЩЗжЮЊСНДѓЯЕСаЃЌЭЈЪНЗжБ№ЮЊЃКBnHn+6(B4H10)ЃЌBnHn+4 (B2H6ЃЌB5H9ЃЌB6H10)ЃЌГЦЮЊХ№ЭщЁЃзюМђЕЅЕФХ№ЭщЪЧввХ№ЭщЃЈB2H6ЃЉЁЃ
Х№ЧтЛЏКЯЮяЕФЗжзгНсЙЙВЛФмНігУвЛАуЕФЙВМлМќРДБэЪОЁЃвђЮЊХ№дзгЪЧШБЕчзгдзгЃЌХ№ЭщЗжзгФкЫљгаМлЕчзгЕФЪ§ВЛФмТњзувЛАуЙВМлМќЫљашвЊЕФЪ§ФПЁЃдкB2H6ЗжзгжаЃЌЙВга14ИіМлЙьЕРЃЌЖјжЛга12ИіМлЕчзгЃЌЫљвдB2H6ЪЧШБЕчзгЛЏКЯЮяЁЃЁЁ
ТБЫиЖМФмКЭХ№аЮГЩХ№ЕФТБЛЏЮяЃЌМДШ§ТБЛЏХ№ЁЃ
жЦБИЃКЭЈГЃПЩгУХ№гыТБЫижБНгЗДгІжЦЕУШ§ТБЛЏХ№ЃК
2B+3X2 Ёњ 2BX3
вВПЩвдгЉЪЏЃЌХЈH2SO4КЭB2O3ЗДгІжЦБИBF3ЃК
B2O3+3CaF2+3 H2SO4Ёњ 2BF3+3CaSO4+3H2O
гУB2O3гыHFЫсзїгУЃЌПЩжЦЕУBF3ЃК
B2O3+6HFЁњ 2BF3+3H2O
гУжУЛЛЗЈЃЌЪЙBF3гыAlCl3ЛђAlBr3ЗДгІЃЌПЩЕУBCl3ЛђBBr3ЃК
BF3(g)+ AlCl3 Ёњ AlF3+ BCl3
BF3(g)+ AlBr3 Ёњ AlF3+ BBr3
гУТБЛЏЗЈЃЌвдB2O3КЭCЮЊдСЯ ЃЌЭЈШыCl2ЦјЃЌ,вВПЩжЦБИBCl3ЃК
B2O3+3C+3Cl2 Ёњ 2BCl3+3CO
НсЙЙЃКШ§ТБЛЏХ№ЕФЗжзгНсЙЙЖМЪЧЦНУцШ§НЧаЮ(ШчЯТЭМ)ЃЌБэУїBдзгЖМЪЧвдsp2дгЛЏЙьЕРгыТБ дзгаЮГЩІвМќЁЃ
дзгаЮГЩІвМќЁЃ
![]()
аджЪЃКШ§ТБЛЏХ№ЖМЪЧЙВМлЛЏКЯЮяЃЌШлЉpЗаЕуОљКмЕЭЃЌВЂгаЙцТЩЕиАДFЉpClЉpBЉpIЫГађЖјж№НЅдіИпЃЌЫќУЧЕФЛг ЗЂадЫцЯрЖдЗжзгжЪСПЕФдіДѓЖјНЕЕЭЁЃ
BF3ЪЧЮоЩЋЕФгажЯЯЂЦјЮЖЕФЦјЬхЃЌВЛФмШМЩеЃЌBF3ЫЎНтЕУЕНЗњХ№ЫсH[BF4]ЃК
4BF3+3H2O Ёњ 3H[BF4]+H3BO3
ЬхЯжГіBF3ЪЧШБЕчзгЛЏКЯЮяЃЌЪЧКмЧПЕФLewisЫсЁЃЗњХ№ЫсЪЧЧПЫсЃЌНівдРызгзДЬЌДцдкгкЫЎШмвКжаЁЃ
BCl3ТдМгбЙСІМДПЩвКЛЏЃЌЫќЪЧЮоЩЋОпгаИпелЩфТЪЕФвКЬхЁЃдкГБЪЊЕФПеЦјжаЗЂбЬВЂдкЫЎжаЧПСвЫЎНтЃК
BCl3+3H2O Ёњ H3BO3+3HCl
4. ЕЊЛЏХ№
ЕЊЛЏХ№ЪЧвЛжжаТаЭЕФЮоЛњКЯГЩВФСЯЁЃдк873KЪБЃЌ B2O3гыNH3ЗДгІПЩжЦЕУЕЊЛЏХ№BNЁЃBNгыO2ЪЧЕШЕчзгЬхЁЃЕШЕчзгЬхГЃБэЯжГіЯрЫЦЕФНсЙЙКЭЯрЫЦЕФаджЪЁЃ
BNгаШ§жжОЇаЭЃК
ЂХЮоЖЈаЭ(РрЫЦгкЮоЖЈаЭЬП)
ЂЦСљЗНОЇаЭ(РрЫЦгкЪЏФЋ)ЫќгжГЦЮЊАзЪЏФЋЃЌЪЧвЛжжгХСМЕФФЭИпЮТШѓЛЌМСЁЃ
ЂЧСЂЗНОЇаЭ(РрЫЦгкН№ИеЪЏ)ЃЌгВЖШШчН№ИеЪЏЃЌгУзїФЅСЯЁЃ
12.3.4 ТСЕФЛЏКЯЮя
ТСЪЧЕфаЭЕФСНаддЊЫиЃЌТСЕФЕЅжЪКЭЛЏКЯЮяМШФмШмгкЫсгжФмШмгкМюЖјГЪЯжЕфаЭЕФСНадЁЃ
1. бѕЛЏТСКЭЧтбѕЛЏТС
бѕЛЏТСAl2O3гаЖржжОЇаЭЃЌЦфжаСНжжжївЊБфЬхЪЧІС-Al2O3КЭІУ-Al2O3ЁЃ
ІС-Al2O3ГЦЮЊИегёЃЌШлЕуИпЃЌгВЖШДѓЃЌВЛШмгкЫсЁЂМюЁЃ
ІУ-Al2O3ГЦЮЊЛюадбѕЛЏТСЃЌПЩШмгкЫсЁЂМюЃЌПЩгУзїДпЛЏМСдиЬхЁЃ
гааЉбѕЛЏТСдиЬхЛљБОЩЯЪЧЭИУїЕФЃЌвђКЌгаЩйСПдгжЪЖјГЪЯжЯЪУїЕФбеЩЋЁЃКьБІЪЏКЌгаМЋЮЂСПИѕЕФбѕЛЏЮяЃЌРЖБІЪЏКЌгаЬњКЭюбЕФбѕЛЏЮяЃЌЛЦОЇКЌгаЬњЕФбѕЛЏЮяЁЃ
КьБІЪЏ(Cr3+) РЖБІЪЏ(Fe3+ЃЌCr3+) ЛЦгё/ЛЦОЇ(Fe3+)
ЧтбѕЛЏТСЪЧСНадбѕЛЏЮяЃЌЫќШмгкЫсЩњГЩ ![]() ЁЃ
ЁЃ
2.ТСЕФТБЛЏЮя
РызгаЭЛЏКЯЮя | AlF3 |
|
|
ЙВМлаЭЛЏКЯЮя | AlCl3 | AlBr3 | AlI3 |
ЙВМлаЭЛЏКЯЮяЕФЙВЭЌЬиЕуЃКШлЕуЕЭЃЌвзЛгЗЂЃЌвзШмгкгаЛњШмМСЃЌвзаЮГЩЫЋОлЮяЁЃ
ЮоЫЎAlCl3ЕФЫЎНтЗДгІЗЧГЃОчСвВЂЗХГіДѓСПЕФШШЃЌЩѕжСдкГБЪЊЕФПеЦјжавВвђЧПСвЕФЫЎНтЖјЗЂбЬЁЃ
3.ТСЕФКЌбѕЫсбЮ
ГЃМћЕФТСЕФКЌбѕЫсбЮгаСђЫсТСAl2(SO4)3ЃЌТСМиЗА(УїЗЏ)KAl(SO4)2ЁЄ12H2OЕШЁЃ
СђЫсТСЁЂЯѕЫсТСШмгкЫЎКѓЃЌгЩгкAl3+ЕФЫЎНтЃЌШмвКГЪЫсадЁЃ
![]()
ТСЕФШѕЫсбЮЫЎНтИќМгУїЯдЃЌЩѕжСДяЕНМИКѕЭъШЋЕФГЬЖШЁЃ
![]()
![]()
ЫљвдЃЌШѕЫсЕФТСбЮВЛФмгУЪЊЗЈжЦЕУЁЃ
Al3+ЕФМјЖЈЃКдкАБМюадЬѕМўЯТЃЌМгШымчЫиЃЌЩњГЩКьЩЋГСЕэЁЃ

Ёь12.4ЁЁpЧјдЊЫиЛЏКЯЮяаджЪЕФЕнБфЙцТЩ
12.4.1 pЧјдЊЫиЕФЧтЛЏЮя
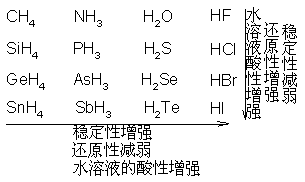
12.4.2 pЧјдЊЫиЕФбѕЛЏЮяМАЦфЫЎКЯЮя
1.бѕЛЏЮя
pЧјдЊЫиДѓЖрЪ§ЖМФмгыбѕжБНгЛђМфНгЕиаЮГЩбѕЛЏЮяЁЃЯЁгаЦјЬхжажЛгаыЏФмМфНгЕигыбѕаЮГЩбѕЛЏЮяЁЃЭЌзхдЊЫиЭЌвЛбѕЛЏжЕЕФбѕЛЏЮяЕФЫсМюадБфЛЏЙцТЩздЩЯЖјЯТЫсадж№НЅМѕШѕЃЌМюадж№НЅдіЧПЁЃ
ЂВбѕЛЏЮяЫЎКЯЮяЕФЫсМюад
ЭЌвЛжмЦкpЧјдЊЫизюИпбѕЛЏжЕбѕЛЏЮяЕФЫЎКЯЮяДгзѓЕНгвМюадМѕШѕЃЌЫсаддіЧПЁЃР§ШчЃК
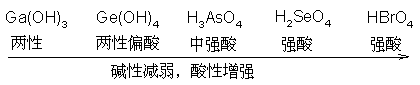
ЭЌзхдЊЫиЯрЭЌбѕЛЏжЕбѕЛЏЮяЕФЫЎКЭЮяЕФЫсМюадздЩЯЖјЯТЫсадМѕШѕЃЌМюаддіЧПЁЃ
LЁЄPaulingдкбаОПКЌбѕЫсЧПЖШгыНсЙЙжЎМфЕФЙиЯЕЪБЃЌзмНсГіЯТУцЕФСНЬѕОбщЙцдђЁЃ
LЁЄPaulingЙцдђ(ЖЈад)
Ёя ЧтбѕЛЏЮяЛђКЌбѕЫсЃЌПЩМЧзїЃК(HO)mROn,Цфжаm:єЧЛљбѕЕФИіЪ§ЃЛnЃКЗЧєЧЛљбѕЕФИіЪ§ЁЃЫсадЕФЧПШѕШЁОігкєЧЛљЧтЕФЪЭЗХФбвзЃЌЖјєЧЛљЧтЕФЪЭЗХгжШЁОігкєЧЛљбѕЕФЕчзгУмЖШЃЌШєєЧЛљбѕЕФЕчзгУмЖШаЁЃЌвзЪЭЗХЧтЃЌдђЫсадЧПЁЃ
Ёя ШєжааФдзгRЕФЕчИКадДѓЃЌАыОЖаЁЃЌбѕЛЏжЕИпдђєЧЛљбѕЕФЕчзгУмЖШаЁЃЌЫсадЧПЃЛЗЧєЧЛљбѕЕФЪ§ФПЖрЃЌПЩЪЙєЧЛљбѕЩЯЕФЕчзгУмЖШМѕаЁЃЌЫсаддіЧПЁЃР§ШчЃК
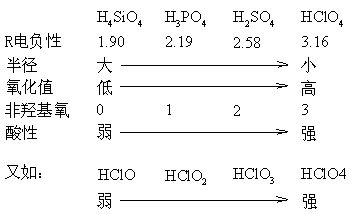
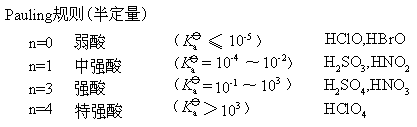
ЂГ pЧјдЊЫиЛЏКЯЮяЕФбѕЛЏЛЙдад
ЖдгкЭЌвЛдЊЫиВЛЭЌбѕЛЏжЕЕФЛЏКЯЮяРДЫЕЃЌзюИпбѕЛЏжЕЕФЛЏКЯЮяжЛПЩФмгабѕЛЏадЃЌЧвЭЌвЛжмЦкЕФзюИпбѕЛЏжЕЛЏКЯЮяЕФбѕЛЏадДгзѓЕНгввРДЮдіЧПЁЃР§ШчЃК
ЕчЖд | Al3+/Al | SiO2/Si | H3PO4/P |
|
|
| -1.68 | -0.9754 | 0.412 | 0.384 | 1.390 |
ЖдгкТШЁЂфхЁЂЕтЕШЗЧН№ЪєадНЯЧПЕФдЊЫиЕФВЛЭЌбѕЛЏжЕЕФКЌбѕЫсРДЫЕЃЌЭЈГЃВЛЮШЖЈЕФЫсбѕЛЏадНЯЧПЁЃР§ШчЃК
ЕчЖд |
|
| HClO2/Cl- | HClO/Cl- |
| 1.38 | 1.45 | 1.57 | 1.49 |
ЂД pЧјдЊЫиКЌбѕЫсЕФШШЮШЖЈад
вЛАуЕиЫЕЃЌКЌбѕЫсЕФШШЮШЖЈадВюЃЌдђЯргІЕФбЮЕФШШЮШЖЈадвВНЯВюЁЃЕЋЪЧЃЌКЌбѕЫсбЮБШЯргІЕФКЌбѕЫсвЊЮШЖЈаЉЁЃ
ЭЌвЛжжКЌбѕЫсаЮГЩЕФбЮЃЌвЛАуРДЫЕЃЌН№ЪєдНЛюЦУЃЌЯргІЕФКЌбѕЫсбЮвВдНЮШЖЈЁЃР§ШчЃК
| Na2CO3 | BaCO3 | MgCO3 | FeCO3 | CdCO3 | Ag2CO3 |
ЗжНтЮТЖШ/Ёц | 1800 | 1360 | 540 | 280 | 345 | 275 |
ЕкЪЎШ§еТ Й§ЖЩдЊЫи(вЛ)
БОеТЮвУЧжївЊСЫНтЙ§ЖЩдЊЫиЕФЭЈадЁЃСЫНтюбЁЂЗАМАЦфживЊЛЏКЯЮяЕФаджЪЁЃ ЪьЯЄИѕЕФЕчЪЦЭМЃЌеЦЮе Cr(Ђѓ ),Cr( Ђі)ЛЏКЯЮяЕФЫсМюадЁЂбѕЛЏЛЙдадМАЦфЯрЛЅзЊЛЏЁЃСЫНтютЮйЕФЛЏКЯЮяЁЃ ЪьЯЄУЬЕФЕчЪЦЭМЃЌеЦЮе Mn(Ђђ ),Mn( Ђє),Mn(Ђі),Mn(Ђї)ЕФживЊЛЏКЯЮяЕФаджЪКЭЗДгІЁЃеЦЮеFe(Ђђ ),Co( Ђђ),Ni(Ђђ)ЕФживЊЛЏКЯЮяЕФаджЪМАЦфБфЛЏЙцТЩЃЛеЦЮе Fe(Ђѓ ),Co( Ђѓ),Ni(Ђѓ)ЕФживЊЛЏКЯЮяЕФаджЪМАЦфБфЛЏЙцТЩЁЃЪьЯЄЬњЁЂюмЁЂФјЕФживЊХфКЯЮяЁЃ
Ёь13.1 dЧјдЊЫиИХЪі
13.1.1 dЧјдЊЫиМђНщ
dЧјдЊЫиАќРЈжмЦкЯЕЕкЂѓBЁЋЂї B,Ђї,Ђё BЁЋЂђBдЊЫиЃЌВЛАќРЈячЯЕКЭяЙЯЕдЊЫиЁЃ dЧјдЊЫиЖМЪЧН№ЪєдЊЫиЁЃdЧјдЊЫиЕФМлЕчзгЙЙаЭЮЊЃЌ(n-1)d1-10ns1-2(PdЮЊ5s0)ЁЃЭЌжмЦкdЧјдЊЫиН№ЪєадЕнБфВЛУїЯдЃЌЭЈГЃАДВЛЭЌжмЦкНЋЙ§ЖЩдЊЫиЗжЮЊШ§ИіЙ§ЖЩЯЕЃК
ЕквЛЙ§ЖЩЯЕЃКЕкЫФжмЦкдЊЫиДгюж(Sc)ЕНаПZn;
ЕкЖўЙ§ЖЩЯЕЃКЕкЮхжмЦкдЊЫиДгюЦ(Y)ЕНяг(Cd);
ЕкШ§Й§ЖЩЯЕЃКЕкСљжмЦкдЊЫиДгях(Lu)ЕНЙЏ(Hg)ЁЃ
dЧјдЊЫидкздШЛНчжаДЂСПвдЕквЛЙ§ЖЩЯЕЮЊНЯЖрЃЌЫќУЧЕФЕЅжЪКЭЛЏКЯЮядкЙЄвЕЩЯЕФгУЭОвВНЯЙуЗКЁЃБОеТНЋжиЕубЇЯАДгюб(Ti)ЕНФј(Ni)ет7ИідЊЫиЃЌЛЙвЊЪЪЕБСЫНтЮвЙњЕФЗсВњдЊЫиют (Mo)КЭЮй(W)ЁЃ
13.1.2 dЧјдЊЫиЕФдзгАыОЖКЭЕчРыФм
ЭЌжмЦкЙ§ЖЩдЊЫиЕФдзгАыОЖЫцзХдзгађЪ§ЕФдіМгЖјЛКТ§ЕивРДЮМѕаЁЃЌЕНСЫЕкЂјзхдЊЫиКѓгжЛКТ§діДѓЁЃЭЌзхЙ§ЖЩдЊЫиЕФдзгАыОЖЃЌГ§СЫЂѓBЭтЃЌздЩЯЖјЯТЫцзХдзгађЪ§ЕФдіДѓЖјдіДѓЁЃ
ИїЙ§ЖЩЯЕдЊЫиЕчРыФмЫцдзгађЪ§ЕФдіДѓЃЌзмЕФБфЛЏЧїЪЦЪЧж№НЅдіДѓЕФЁЃЭЌИБзхЙ§ЖЩдЊЫиЕФЕчРыФмЕнБфВЛКмЙцдђЁЃdЧјдЊЫиЕФЕквЛЕчРыФмБфЛЏЧїЪЦШчЭМЫљЪОЁЃ
13.1.3 dЧјдЊЫиЕФЮяРэаджЪ
Ёя ШлЕуЁЂЗаЕуИпЁЃШлЕузюИпЕФЕЅжЪЪЧЮй(W)
Ёя гВЖШДѓЁЃгВЖШзюДѓЕФН№ЪєЪЧИѕ(Cr)
Ёя УмЖШДѓЁЃУмЖШзюДѓЕФЕЅжЪЪЧяА(Os)
Ёя ЕМЕчадЁЂЕМШШадЁЂбгеЙадКУЁЃ
13.1.4 dЧјдЊЫиЕФЛЏбЇаджЪ
дкЛЏбЇаджЪЗНУцЃЌЕквЛЙ§ЖЩЯЕдЊЫиЕФЕЅжЪБШЕкЖўЁЂШ§Й§ЖЩЯЕдЊЫиЕФЕЅжЪЛюЦУЁЃЛЏбЇаджЪБфЛЏзмЧїЪЦЪЧЭЌвЛЙ§ЖЩЯЕЕЅжЪЕФЛюЦУадДгзѓЕНгвНЕЕЭЁЃ
13.1.5 dЧјдЊЫиЕФбѕЛЏЬЌ
Й§ЖЩдЊЫиДѓЖрПЩвдаЮГЩЖржжбѕЛЏжЕЕФЛЏКЯЮяЁЃЭМжаИјГіСЫЕквЛЙ§ЖЩЯЕдЊЫиЕФИїжжбѕЛЏжЕЃЌКьЩЋДњБэГЃМћЕФбѕЛЏжЕ
13.1.6 dЧјдЊЫиЕФРызгЕФбеЩЋ
Й§ЖЩдЊЫиЕФЫЎКЯРызгДѓЖрЪЧгабеЩЋЕФЁЃ
Й§ЖЩдЊЫигыЦфЫќХфЬхаЮГЩЕФХфРызгвВГЃОпгабеЩЋЁЃетаЉХфРызгЮќЪеСЫПЩМћЙт(>30nmЁЋ 400nm)ЕФвЛВПЗжЗЂЩњСЫdЁЊdдОЧЈЃЌЖјАбЦфгрВПЗжЕФЙтЭИЙ§ЛђЩЂЩфГіРДЃЌЮвУЧПДЕНЕФЮяжЪЕФбеЩЋОЭЪЧетВПЗжЭИЩфЙтЛђЩЂЩфЙтЁЃ
ЖдгкФГаЉОпгабеЩЋЕФКЌбѕЫсИљРызгЃЌШч (ЛЦЩЋ), (зЯЩЋ)ЕШЃЌЫќУЧЕФбеЩЋБЛШЯЮЊЪЧгЩЕчКЩЧЈвЦв§Ц№ЕФЁЃ
Ёь13.2.юбИБзх Ti, Zr, HfЁЁЁЁ ЁЁ
вЛ.юбЕФДцдкгывБСЖ
- ЯЁгадЊЫи
TiЪєгкЯЁгадЊЫиЃЌTiгаИЛПѓКьН№ЪЏ(TiO2)КЭюбЬњПѓ(FeTiO3)ЃЌдкЕиПЧжаКЌСПЮЊ0.42%, ОгЕк10ЮЛЁЃ
Zr вдяЏЪЏЕШаЮЪНДцдк, Ог20ЮЛ, БШЪьжЊЕФCuКЭZnЖМЖрЁЃ
2. юбЕФвБСЖ
ЁЁЁЁЁЁЁЁЙЄвЕЩЯвдюбЬњПѓЮЊдСЯЃЌжЦШЁюбЕЅжЪЁЃ
![]()
ЁЁЁЁЯШгУХЈH2SO4ДІРэФЅЫщЕФюбЬњПѓЗл:
ЁЁЁЁFeTiO3 + 3H2SO4 ===Ti(SO4)2 + FeSO4 + 3H2O
ЁЁЁЁПѓЪЏжаЕФFeOКЭFe2O3вВЭЌЪБзЊБфГЩСЫСђЫсбЮЃЌМгШыFeЗлЃЌЛЙдFe2(SO4)3жСFeSO4ЃЌРфШДЪЙFeSO4ЁЄ7H2O(ТЬЗЏ)НсОЇЃЌЕУИБВњЦЗЁЃ
ЁЁЁЁЫЎНтTi(SO4)2:
ЁЁЁЁTi(SO4)2 + H2O=== TiOSO4(СђЫсбѕюб) + H2SO4
ЁЁЁЁTiOSO4 + 2H2O ===H2TiO3(ГСЕэАзЩЋЃЌЦЋюбЫс) + H2SO4
ЁЁЁЁьбЩеH2TiO3жЦЕУTiO2
ЁЁЁЁЁЁH2TiO3 ![]() TiO2 + H2O
TiO2 + H2O
ЁЁЁЁТШЛЏЁЂёюКЯжЦTiCl4
ЁЁЁЁЁЁ TiO2 + 2C + 2Cl2 ![]() TiCl4(l) + 2CO(ЦјЬх)
TiCl4(l) + 2CO(ЦјЬх)
дкArЦјЗеБЃЛЄЯТЃЌгУШлУОЛЙдTiCl4еєЦј
ЁЁЁЁЁЁ TiCl4(g) + 2Mg(l) --- Ti + 2MgCl2(l) (ШлШк, ArЦјБЃЛЄ)
ПЩвдНЋЪЃгрЕФMgКЭЩњГЩЕФMgCl2еєЗЂЕєЃЌЛђгУбЮЫсНЋMgКЭMgCl2ШмЕєЃЌЕУКЃУрюбдйШлСЖЃЌЕУTiЕЅжЪЁЃ вВПЩжБНгТШЛЏН№КьЪЏПѓЗлЃЌжЦTiCl4ЃЌЭъГЩюбЕФвБСЖЁЃ
Жў юбЕЅжЪ
1. ЮяРэаджЪ
ЁЁЁЁвјАзЩЋЃЌУмЖШ4.54ЃЌБШИжЬњЕФ7.8аЁЃЌБШТСЕФ2.7ДѓЃЌНЯЧсЃЌЧПЖШНгНќИжЬњЃЌМцгаТСЬњЕФгХЕуЃЌМШЧсЧПЖШгжИп(КНЬьВФСЯ, блОЕМмЕШ)ЁЃ
ЁЁЁЁжЦзїМЧвфадКЯН№NTКЯН№(ФјюбКЯН№, NT)ЃЌ
ЁЁЁЁЧзЩњЮяадЃЌШЫдьЙЧ, ПЩгыЩњЮяЬхЙЧШтвзГЄдквЛЦ№ЁЃ
- ЛЏбЇаджЪ
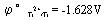
ЁЁЁЁШШСІбЇЩЯКмЛюЦУЃЌЕЋБэУцЖлЛЏЃЌдкГЃЮТЯТМЋЮШЖЈЁЃГЃЮТВЛгыX2ЁЂO2ЁЂH2OЗДгІЃЌВЛгыЧПЫс(АќРЈЭѕЫЎ)ЃЌвдМАЧПМюЗДгІЁЃюбКЯН№ФЭЫсМюИЏЪДЁЃЕЋИпЮТЪБюбЯрЕБЛюЦУ:
ЁЁЁЁTi + O2 === TiO2 (КьШШ)
ЁЁЁЁTi + 2Cl2 === TiCl4 (600K)
ЁЁЁЁ3Ti + 2N2 === Ti3N4 (800K)
ЁЁЁЁTiЕФвдЩЯЗДгІОљДяЕНзюИпбѕЛЏЬЌЁЃ
ЁЁЁЁ2Ti + 6HCl ==2TiCl3(зЯЩЋ) + 3H2ЃЈЦјЬхЃЉ
ЁЁЁЁзюКУЕФШмМСЪЧЧтЗњЫсЛђЧтЗњЫсгыбЮЫсЕФЛьКЯвКЃЌВњЩњ![]() :
:
ЁЁЁЁTi + 6HF ===TiF62- + 2H+ + 2H2ЃЈЦјЬхЃЉ
ЁЁЁЁTiВЛШмгкШШМюЃЌЕЋКЭШлШкМюзїгУ:
ЁЁЁЁ2Ti + 6KOH ====2K3TiO3 + 3H2 (ШлШк)
ЁЁЁЁзмжЎ, юбУмЖШаЁЃЌЧПЖШИпЃЌПЙЫсМюИЏЪДЃЌгаМЧвфадЃЌЧзЩњЮяадЃЌдкЕиПЧжаДЂСПИпЃЌЪЧМЋгаЧАЭОЕФНсЙЙВФСЯЃЌБЛгўЮЊЁАЕкШ§Н№ЪєЁБКЭЁАЖўЪЎвЛЪРМЭН№ЪєЁБЁЃ
Ш§ юбЕФживЊЛЏКЯЮя
1. ЖўбѕЛЏюб
1ЁугыЫсЕФзїгУ
ЁЁЁЁTiO2АзЩЋЗлФЉЃЌЫзГЦюбАзЃЌВЛШмгкH2OЁЂЯЁЫсКЭЯЁМюжаЃЌдквЛЖЈЕФЬѕМўЯТПЩШмгкШШХЈH2SO4жаЁЃ
ЁЁЁЁTiO2 + 2H2SO4(ХЈ) ![]() Ti(SO4)2 + 2H2O
Ti(SO4)2 + 2H2O
ЁЁ![]() ЕчГЁЙ§ЧПЃЌдкЫЎжавзЫЎНтГЩ:
ЕчГЁЙ§ЧПЃЌдкЫЎжавзЫЎНтГЩ:
ЁЁЁЁ![]()
ЁЁ![]() ГЦЮЊюббѕЛљЛђюбѕЃЛљЃЌвђДЫЩЯЪіЗДгІПЩаДГЩ:
ГЦЮЊюббѕЛљЛђюбѕЃЛљЃЌвђДЫЩЯЪіЗДгІПЩаДГЩ:
ЁЁЁЁTiO2 + H2SO4(ХЈ) ![]() TiOSO4 + H2O
TiOSO4 + H2O
ЁЁ![]() КЭ
КЭ![]() жЎМфгаШчЯТЦНКт:
жЎМфгаШчЯТЦНКт:
ЁЁЁЁ![]()
ЫЎШмвКжаВЛФмЮіГі Ti(SO4)2ЃЌШДПЩвдЮіГіАзЩЋЗлФЉTiOSO4ЁЄH2OЁЃ
TiO2гыKHSO4ЙВШлЃЌЕУПЩШмадбЮРр:
ЁЁЁЁTiO2 + 2KHSO4 ===TiOSO4 + K2SO4 + H2O ЃЈШлШкЃЉ
2ЁугыМюадЛЏКЯЮязїгУ
ЁЁЁЁЁЁTiO2 + MgO=== MgTiO3 (ШлШк)
ЁЁЁЁЁЁTiO2 + BaCO3=== BaTiO3 + CO2(ЦјЬх) ЃЈШлШкЃЉ
ЁЁЁЁЁЁBaTiO3ЃЌЦЋюбЫсБЕЃЌЪЧвЛжжбЙЕчВФСЯЃЌЪмбЙЪБСНЖЫВњЩњЕчЮЛВюЁЃ
2. юбЫс
ЁЁЁЁдкюббЮжаМгМюЃЌПЩЕУ![]() -юбЫс(ЧтбѕЛЏюб):
-юбЫс(ЧтбѕЛЏюб):
ЁЁЁЁЁЁTiBr4 + 4NaOH === Ti(OH)4(ГСЕэ) + 4NaBr![]() -юбЫсЛюадДѓЃЌПЩШмгкЫсЛђМюЃЌПЩвдаДГЩTi(OH)4ЁЂH4TiO4ЛђTiO2ЁЄxH2OЕШаЮЪНЁЃ
-юбЫсЛюадДѓЃЌПЩШмгкЫсЛђМюЃЌПЩвдаДГЩTi(OH)4ЁЂH4TiO4ЛђTiO2ЁЄxH2OЕШаЮЪНЁЃ
НЋюбЫсШмвКжѓЗаЃЌЫЎНтЩњГЩ![]() -юбЫсЃЌетжжЫЎНтМДЪЙМгЧПЫсвВВЛФмвжжЦЁЃ
-юбЫсЃЌетжжЫЎНтМДЪЙМгЧПЫсвВВЛФмвжжЦЁЃ
ЁЁЁЁTi(SO4)2 + 4H2O![]() Ti(OH)4 + 2H2SO4
Ti(OH)4 + 2H2SO4
ЁЁЁЁЕУЕНЕФ![]() -юбЫсЮШЖЈЃЌВЛШмгкЫсвВВЛШмгкМюЁЃ
-юбЫсЮШЖЈЃЌВЛШмгкЫсвВВЛШмгкМюЁЃ
3. ЫФТШЛЏюб
ЁЁЁЁTiCl4ЮоЩЋШмвКЃЌгаДЬМЄадЦјЮЖЃЌМЋвзЫЎНтЃЌдкПеЦјжаУААзбЬ
ЁЁЁЁЁЁЁЁTiCl4 + H2O === H2TiO3 + 4HCl
ЁЁЁЁЁЁЁЁжЦTiCl4ЙиМќЪЧЗРжЙЫЎНтЁЃ
4. Ш§ТШЛЏюб
ЁЁЁЁЕЅжЪюбдкМгШШЧщПіЯТгыбЮЫсЗДгІЕУTiCl3зЯЩЋШмвКЁЃ
ЁЁЁЁTiCl3вВПЩвдгЩTiCl4ЛЙджЦЕУ:
ЁЁЁЁЁЁ2TiCl4 + H2 ===TiCl3 + 2HCl
ЁЁЁЁЁЁ2TiCl4 + Zn === TiCl3 + ZnCl2
ЁЁЁЁДгЫЎШмвКжаПЩвдЮіГіTiCl3ЁЄ6H2OЕФзЯЩЋОЇЬхЃЌХфКЯЮяЕФЙЙГЩЪЧ[Ti(H2O)6]Cl3ЁЃ ШєгУввУбДгTiCl3ЕФБЅКЭШмвКжанЭШЁГіЃЌПЩЕУTiCl3 ЁЄ6H2OТЬЩЋОЇЬхЃЌХфКЯЮяЕФЙЙГЩЪЧ[Ti(H2O)5Cl]Cl2ЁЄH2OЁЃСНепЛЅЮЊЫЎКЯвьЙЙЁЃ
ЁЁЁЁгаЙиTi(IV)КЭTi(III)ЕФЕчМЋЕчЪЦШчЯТЃК
ЁЁЁЁ![]()
ЁЁЁЁПЩМћЫЕВЛЩЯTi(IV)ЕФбѕЛЏадЃЌЕЙПЩЬИTi(III)ЕФЛЙдад
![]() ЁЁ
ЁЁ![]()
ЫФ яЏгыюў
гЩгкячЯЕЪеЫѕЕФгАЯьЃЌZr гыHf МЋЮЊЯрЫЦЃЌОљЮЊЧГЛвЩЋКЭЛвЩЋН№ЪєЁЃдЊЫи Zr ЗЂЯжКѓЃЌДѓдМ 30 ФъвдКѓЗЂЯж HfЃЌВЂЗЂЯж 30 ФъРДЖдZr НјааЕФбаОПЃЌШЋЪЧдкКЌ 2ЃЅ Hf ЕФЛљДЁЩЯЭъГЩЕФЁЃ
Zr КЭ Hf ФЭЫсадБШ Ti ЛЙЧПЃЌгШЦфЪЧ HfЃЌ100Ёц вдЯТЖдЫсЮШЖЈ(HF Г§Эт )ЃЌгыМюПЩЗДгІ:
ЁЁЁЁЁЁЁЁZr + 4KOH === K4ZrO4 + 2H2(ЦјЬх)
ЁЁЁЁZrO2 ЪЧАзЩЋЗлФЉЃЌгВЖШИпЃЌИпЮТДІРэЕФ ZrO2, Г§ HF Эт, ВЛШмгкЦфЫќЫсЁЃ ГЃгУЕФПЩШмадяЏЪЧ ZrOCl2ЃЌвзЫЎНт
ZrOCl2 + (x+1)H2O === ZrO2ЁЄxH2O(яЏЫс) + 2HCl
ЁЁЁЁяЏЫсБШюбЫсШѕЃЌвВгаСНадЁЃ ЁЁ
Ёь13.2.ЗАИБзхЁЁЁЁ ЁЁ
ЁЁвЛ.ЗАЕЅжЪ
ЗАдкздШЛНчжаЗЧГЃЗжЩЂЃЌV(III)ОГЃКЭЬњПѓЛьЩњЃЌШчЗАюбЬњПѓЃЌV(V)ЩаПЩЖРСЂГЩПѓЃЌЗАЪЧЯЁгаН№ЪєЁЃ
ЗАЧГЛвЩЋЃЌИпШлЕу(БШTiИп)ЃЌДПОЛЪБбгеЙадКУЃЌВЛДПЪБгВЖјДрЁЃИжжаМг0.1- 0.2%ЕФЗАЃЌШЭадЁЂЧПЖШЁЂбгеЙадМАПЙГхЛїСІОљМгЧПЁЃ
ЁЁЁЁЁЁЁЁ![]()
ЁЁЁЁШШСІбЇЩЯПДЃЌгІЪЧМЋЛюЦУЕФН№ЪєЃЌЕЋгЩгкБэУцЖлЛЏЃЌГЃЮТЯТВЛЛюЦУЃЌПщзДЕФЗАПЩвдЕжПЙПеЦјЕФбѕЛЏКЭКЃЫЎЕФИЏЪДЃЌЗЧбѕЛЏадЫсМАМювВВЛФмгыЗАзїгУЁЃ
ЁЁЁЁVПЩвдШмгкХЈСђЫсКЭЯѕЫсжа
ЁЁЁЁЁЁV + 8HNO3 == V(NO3)4 + 4NO2 + 4H2O
ЁЁЁЁЁЁV(NO3)4 ЛђвдVO(NO3)2аЮЪНДцдкЁЃ
ЁЁЁЁИпЮТЯТЛюадКмИп
ЁЁЁЁЁЁЁЁ2V + 5O2 ===V2O5 (зЉКьЩЋЙЬЬх)
ЁЁЁЁЁЁЁЁV + 2Cl2 === VCl4 (КьЩЋвКЬх)
ЁЁЁЁVПЩвдгыШлШкЕФЧПМюЗДгІЁЃ
Жў ЗАЕФживЊЛЏКЯЮя ЁЁ
1. ЮхбѕЛЏЖўЗА
ЁЁЁЁV2O5 , зЉКьЩЋЙЬЬхЃЌЮоГєЁЂЮоЮЖЁЂгаЖОЃЌЪЧЗАЫс H3VO4 МАЦЋЗАЫс HVO3 ЕФЫсєћЁЃ МгШШЦЋЗАЫсяЇПЩЕУV2O5
ЁЁЁЁЁЁ2NH4VO3 === V2O5 + 2NH3 + H2O
ЁЁЁЁШ§ТШбѕЗАЫЎНтвВЕУV2O5
ЁЁЁЁЁЁ2VOCl3 + 3H2O ===V2O5 + 6HCl
ЁЁЁЁЁЁV2O5 дкH2OжаШмНтЖШКмаЁЃЌЕЋдкЫсжаЁЂМюжаЖМПЩШмЃЌЪЧСНадбѕЛЏЮяЁЃ
ЁЁЁЁЁЁV2O5 + 6NaOH === 2Na3VO4 + 3H2O
ЁЁЁЁЁЁV2O5 + H2SO4 === (VO2)2SO4 + H2O
ЁЁЁЁКЭбЮЫсЕФЗДгІЃЌЗХГіCl2ЃЌV2O5габѕЛЏад
ЁЁЁЁЁЁV2O5 + 6HCl === 2VOCl2 + Cl2 + 3H2O
ЁЁ2. ЗАЫсбЮ
ЁЁЁЁЦЋЗАЫсбЮVO3ЁЅЃЌе§ЗАЫсбЮVO43ЁЅЃЌЖўОлЗАЫсбЮV2O74ЁЅЃЌЖрОлЫс(Hn+2VnO3n+1).
ДцдкаЮЪНгыЬхЯЕЕФpHжЕгаЙиЃЌpHдНДѓОлКЯЖШдНЕЭЃЌpHдНаЁЃЌОлКЯЖШдНИпЁЃ
(1) ЕБpH>13ЪБЃЌвдVO43ЁЅДцдкЃЌpHНЕЕЭЃЌОЖўОлЃЌЫФОлЁЂЮхОл.....ж№НЅЩ§ИпЃЛ
(2) ЕБpHЃН2ЪБЃЌвдV2O5аЮЪНЮіГіЃЛ
(3) ЕБpHЁм1ЪБЃЌвдбєРызгДцдк![]() . (V2O5гыЧПЫсЕФЗДгІВњЮя)
. (V2O5гыЧПЫсЕФЗДгІВњЮя)
3. ИїжжбѕЛЏЬЌЕФЗАРызг
ЁЁ ЁЁ 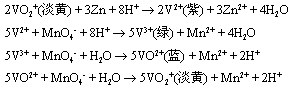
Ш§ юъКЭюу ЁЁ
NbЧГЛвЩЋЃЌTaвјАзЩЋЃЌаджЪЪЎЗжЯрЫЦЁЃNbЁЂTaдкЪвЮТЯТЛюадКмЕЭЃЌгШЦфЪЧTaЃЌЩѕжСВЛКЭЭѕЫЎЗДгІЁЃЕЋПЩвдЛКТ§ЕиШмгкЧтЗњЫсЃЌгШЦфЪЧHF-HNO3ЕФЛьЫсжаЁЃ дкПеЦјжаМгШШОљПЩЕУЮхМлЕФбѕЛЏЮяЃК
ЁЁЁЁЁЁЁЁ4Nb + 5O2 ![]() 2Nb2O5
2Nb2O5
ЁЁЁЁЁЁЁЁ4Ta + 5O2 ![]() 2Ta2O5
2Ta2O5
ЁЁЁЁTa2O5ЯрЕБЮШЖЈЃЌМгШШжС1470ЁцШлЛЏЖјВЛЗжНтЃЌВЛБЛH2ЫљЛЙдЁЃ
ЁЁЁЁNbКЭTaЕФКЌбѕЫсКЭЗАЫсЯрЫЦЃЌгаЖрЫсаЮЪНЃЌNbOCl3взЫЎНт:
ЁЁЁЁЁЁЁЁЁЁ2NbOCl3 + (x+3)H2O=== Nb2O5ЁЄxH2O + 6HCl
Ёь13.3ЁЁИѕ ют Юй ЖрЫсаЭХфКЯЮя
жмЦкЯЕЕкЂі BзхдЊЫивВНаИѕЗжзхЃЌАќРЈИѕ(Cr)ЁЂют(Mo)ЁЂЮй(W)3жждЊЫиЁЃМлЕчзгЙЙаЭЮЊЃК (n-1)d4-5ns1-2ЁЃдкздШЛНчжаЕФжївЊПѓЮягаИѕЬњПѓ(Fe(CrO2)2)ЁЂЛдютПѓ(MoS2)ЁЂКкЮйПѓ(MnFeWO4)ЁЂАзЮйПѓ(CaWO4)ЁЃЮвЙњютПѓзЪдДЗсИЛЃЌЮйПѓЕФДЂСПдМеМЪРНчДЂСПЕФвЛАыЁЃ
13.3.1 Иѕ ют ЮйЕФЕЅжЪ
ИѕЁЂютЁЂЮйЖМЪЧЛвАзЩЋН№ЪєЁЃЫќУЧЕФШлЕуКЭЗаЕуИпЁЃдкЭЈГЃЬѕМўЯТЃЌгЩгкИѕЁЂютЁЂЮйБэУцаЮГЩвЛВубѕЛЏФЄЃЌЫќУЧдкПеЦјЛђЫЎжаЖМЯрЕБЮШЖЈЁЃвђДЫГЃдкЬњжЦЦЗЕФБэУцЖЦгавЛВуИѕЃЌПЩЦ№ЕНЗРИЏЁЂУРЛЏЕФзїгУЁЃЪвЮТЯТЃЌЮоБЃЛЄФЄЕФДПИѕФмШмгкЯЁбЮЫсКЭСђЫсШмвКжаЃЌЖјВЛШмгкЯѕЫсКЭСзЫсЁЃютКЭЮйЖМФмШмгкЯѕЫсКЭЧтЗњЫсЕФЛьКЯШмвКжаЁЃ
13.3.2 ИѕЕФЛЏКЯЮя
ИѕдзгЕФМлЕчзгЙЙаЭЮЊЃК3d54s1ЁЃИѕЕФзюИпбѕЛЏжЕЮЊЃЋ6ЁЃИѕвВФмаЮГЩбѕЛЏжЕЮЊ+5,+4,+3,+2,+1,0,-1,-2ЕФЛЏКЯЮяЁЃИѕЕФживЊЛЏКЯЮягаЃКШ§бѕЛЏИѕ(CrO3)АЕКьЩЋОЇЬхЃЌИѕЫсМи(K2CrO4)ЛЦЩЋОЇЬхЃЌжиИѕЫсМи(K2Cr2O7)ГШКьЩЋОЇЬхЃЌШ§бѕЛЏЖўИѕ(Cr2O3)ТЬЩЋЗлФЉЃЌСђЫсМиИѕ(ИѕМиЗАЃЌ(KCr(SO4)2ЁЄ12H2O)АЕзЯЩЋОЇЬхЁЃ
ИѕЕФживЊЛЏКЯЮяЭЈГЃвдK2Cr2O7ЮЊдСЯжЦБИЃЌЖјK2Cr2O7ЪЧгЩИѕЬњПѓНшжњгкМюШмЗЈжЦЕУЕФЃЌжЦБИK2Cr2O7ЕФСїГЬЭМШчЯТЃК
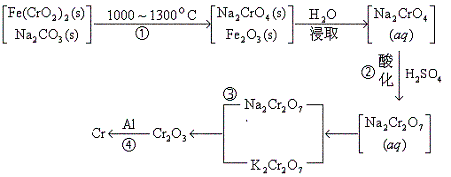
ЫљЩцМАЕНЕФживЊЗДгІЮЊЃК
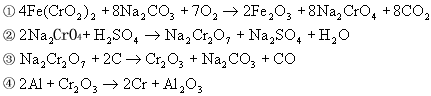
13.3.3 ЫЎШмвКжаИѕЕФРызгМАЦфЗДгІ
1.Cr(Ђѓ)ЕФбЮ
Ёя Cr3+дкЫЎШмвКжаЗЂЩњЫЎНтЁЃ
![]()
дкCr3+ЕФЫЎШмвКжав§ШыШѕЫсИљРызгЃЌЫЎНтНЋНјааЕНЕзЁЃ
2Cr3+ + 3S2Ѓ + 6H2O Ёњ 2Cr(OH)3(s) + 3H2S(g)
2Cr3+ + 3CO32Ѓ + 6H2O Ёњ 2Cr(OH)3(s) + 3H2S(g)
ЯђCr3+ШмвКжаМгШыМюЪБЃЌЯШЩњГЩЛвТЬЩЋЕФ Cr(OH)3ГСЕэЃЌЕБМюЙ§СПЪБЩњГЩССТЬЩЋЕФ ![]() ШмвКЁЃ
ШмвКЁЃ
Ёя Cr(Ђѓ)ОпгаЛЙдадЃЌЕЋдкМюадЬѕМўЯТНЯЧПЃЌМДЃКШєЯыНЋCr(Ђѓ)бѕЛЏЮЊCr(Ђі)ЃЌбЁдёМюадЬѕМўИќШнвзЁЃ
![]()
![]()
![]()
Ёя Cr(Ђѓ)ЕФМјЖЈЃК
дРэСїГЬЃК
![]()
живЊЗДгІЃК
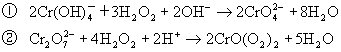
дкЗДгІзюКѓвЛВНМгШыввУбЃЌФПЕФЪЧЪЙCrO(O2)2ФмЙЛНЯЮШЖЈЕиДцдкЁЃ
![]()
Ёя Cr3+гавЛЖЈЕФбѕЛЏад
2Cr3+(зЯЩЋ) + Zn(s) Ёњ 2Cr2+(РЖЩЋ) + Zn2+
Ёя Cr(Ђѓ)ЕФХфКЯЮягаЖржжбеЩЋ
[Cr(H2O)6]Cl3зЯЩЋ, [CrCl(H2O)5]Cl2ЁЄ H2OРЖТЬЩЋ, [CrCl2(H2O)4]ClЁЄ H2OТЬЩЋЁЃ
ЂВCr(Ђі)дкЫЎШмвКжаЕФДцдкМАзЊЛЏ
Ёя pHжЕВЛЭЌЃЌДцдкаЮЪНВЛЭЌ
pHЃМ2ЃЌвд ![]() ЮЊжї,pHЃО6,вд
ЮЊжї,pHЃО6,вд ![]() ЮЊжїЁЃЖўепМфДцдкЯТСаЦНКтЁЃ
ЮЊжїЁЃЖўепМфДцдкЯТСаЦНКтЁЃ
![]()
Ёя ФбШмбЮЕФЩњГЩЪЙ ![]() зЊЛЏЮЊ
зЊЛЏЮЊ ![]()
Р§Шч: ![]()
Ыљвд Яђ ![]() ЕФШмвКжаМгШыAg+ЪБЃЌЩњГЩAg2CrO4
ЕФШмвКжаМгШыAg+ЪБЃЌЩњГЩAg2CrO4
![]()
ЭЌРэЃК

Ёя ![]() ЕФбѕЛЏад
ЕФбѕЛЏад
![]() гаКмЧПЕФбѕЛЏад(
гаКмЧПЕФбѕЛЏад( ![]() ),Жј
),Жј ![]() ЕФбѕЛЏадКмВюЁЃ
ЕФбѕЛЏадКмВюЁЃ
| Fe2+ | Sn2+ |
| H2S | I- | Cl-(ХЈбЮЫсжа) |
ЖдгІВњЮя | Fe3+ | Sn4+ |
| S | I2 | Cl2 |
Р§ШчЃК ![]()
етвЛЗДгІдкЗжЮіЛЏбЇжаГЃгУгкFe2+КЌСПЕФВтЖЈЁЃ
ЂГаЁНс
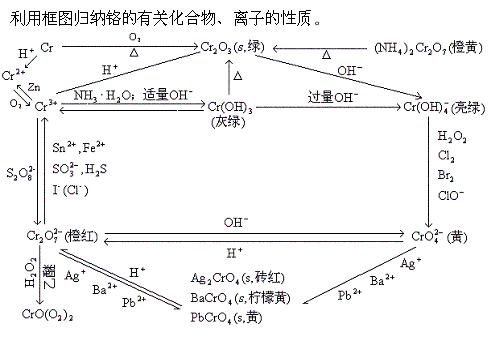
13.3.4 ютЁЂЮйЕФЛЏКЯЮя
ютКЭЮйЕФдзгМлВуЕчзгЙЙаЭЗжБ№ЮЊЃК4d55s1КЭ 5d46s2,ЫќУЧЖМФмаЮГЩбѕЛЏжЕДг+2ЕН+6ЕФЛЏКЯЮяЁЃЦфжабѕЛЏжЕЮЊ+6ЕФЛЏКЯЮяНЯЮШЖЈЁЃР§ШчЃКютЫсяЇ((NH4)2MoO4),ЮйЫсФЦ(Na2WO4)ЕШ.
Ёя МюН№ЪєКЭ ![]() ЕФют(Ђі)ЁЂЮй (Ђі)КЌбѕЫсбЮвзШмгкЫЎЁЃдкПЩШмадЕФЮйЫсбЮЛђютЫсбЮжаЃЌдіМгЫсЖШЭљЭљаЮГЩОлКЯЕФЫсИљРызгЁЃдк(NH4)2MoO4КЭNa2WO4ЕФШмвКжаЗжБ№МгШыЪЪСПЕФбЮЫсЃЌдђЮіГіФбШмгкЫЎЕФютЫсH2MoO4КЭЮйЫсH2WO4ЁЃ
ЕФют(Ђі)ЁЂЮй (Ђі)КЌбѕЫсбЮвзШмгкЫЎЁЃдкПЩШмадЕФЮйЫсбЮЛђютЫсбЮжаЃЌдіМгЫсЖШЭљЭљаЮГЩОлКЯЕФЫсИљРызгЁЃдк(NH4)2MoO4КЭNa2WO4ЕФШмвКжаЗжБ№МгШыЪЪСПЕФбЮЫсЃЌдђЮіГіФбШмгкЫЎЕФютЫсH2MoO4КЭЮйЫсH2WO4ЁЃ
![]()
Ёя ![]() КЭ
КЭ ![]() ПЩвдгыH2SзїгУЃЌЩњГЩСђЛЏЮяЁЃ
ПЩвдгыH2SзїгУЃЌЩњГЩСђЛЏЮяЁЃ
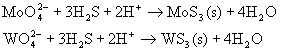
Ёя MoS3КЭWS3ФмШмгк(NH4)2SжааЮГЩСђДњЫсбЮ
![]()
Ёя гУЯѕЫсЫсЛЏЕФютЫсяЇШмвКЃЌМгШШжС50Ёц,дйМгШыNa2HPO4,ЩњГЩСзютЫсяЇ(NH4)3PO4ЁЄ12MoO3ЁЄ 6H2OЛЦЩЋГСЕэ
![]()
етвЛЗДгІГЃгУРД МьВщШмвКжаЪЧЗёКЌга ![]() ,вВПЩгУРДМјЖЈШмвКжаЕФ
,вВПЩгУРДМјЖЈШмвКжаЕФ ![]() ЁЃ
ЁЃ
13.3.5 ЖрЫсаЭХфКЯЮя ЭЌЖрЫсКЭдгЖрЫсМАЦфбЮ
ЖрЫсЃКгЩМђЕЅКЌбѕЫсЫѕКЯЦ№РДЕФЃЌКЌгаИДдгЫсИљРызгЕФКЌбѕЫсЁЃР§ШчЃКH2Cr2O7ЁЃ
ДгХфКЯЮяЕФЙлЕуРДПДЃЌМђЕЅЕФЫсИљРызг ![]() вВЪЧХфРызгЃЌаЮГЩЬхЪЧCr(Ђі),ХфЮЛЬхЪЧO2-ЁЃетРрЫсИљРызгЮЊЕЅКЫХфРызгЁЃЯдШЛЃЌЖрЫсИљ
вВЪЧХфРызгЃЌаЮГЩЬхЪЧCr(Ђі),ХфЮЛЬхЪЧO2-ЁЃетРрЫсИљРызгЮЊЕЅКЫХфРызгЁЃЯдШЛЃЌЖрЫсИљ ![]() ЮЊЖрКЫХфРызгЃЌНсЙЙЮЊЃК
ЮЊЖрКЫХфРызгЃЌНсЙЙЮЊЃК

ЕБаЮГЩЬхЪЧЭЌжжРызгЪБЃЌИУЖрЫсЮЊЭЌЖрЫсЁЃР§ШчЃКH2P2O7ЁЃ
ЕБаЮГЩЬхЪЧВЛЭЌжжРызгЪБЃЌИУЖрЫсЮЊдгЖрЫсЃЌЖдгІЕФбЮЮЊдгЖрЫсбЮЁЃР§ШчЃК (NH4)3PO4ЁЄ 12MO3ЁЄ6H2OЃЛИљОнЪЕбщВтЖЈКЭХфКЯЮяЕФНсЙЙРэТлЃЌАбЫќаДЮЊ (NH4)3[P(Mo3O10)4] ЁЄ6H2OЃЌЦфжаP(Ђѕ)ЪЧаЮГЩЬхЃЌЖјЫФИі ![]() ЪЧХфЮЛЬх
ЪЧХфЮЛЬх
Ёь13.4ЁЁУЬ
жмЦкЯЕЕкЂїBзхдЊЫивВНаУЬзхдЊЫиЃЌАќРЈУЬ(Mn)ЁЂяН(Te)ЁЂяЊ(Re)3жждЊЫиЁЃМлЕчзгЙЙаЭЮЊЃК(n-1)d5ns2ЁЃ
УЬдкЕиПЧжаЕФКЌСПдкЙ§ЖЩдЊЫижаеМЕкШ§ЮЛЃЌНіДЮгкЬњКЭюбЁЃУЬдкздШЛНчжажївЊвдШэУЬПѓMnO2ЁЄxH2OЕФаЮЪНДцдкЁЃ
16.4.1 УЬЕФЕЅжЪ
УЬЪЧАзЩЋН№ЪєЃЌжЪгВЖјДрЃЌЭтаЮгыЬњЯрЫЦЁЃДПУЬгУЭОВЛДѓЃЌГЃвдУЬЬњЕФаЮЪНРДжЦдьИїжжКЯН№ИжЁЃ
Ёя ГЃЮТЯТЃЌУЬФмЛКТ§ЕиШмгкЫЎЃК Mn + 2H2O Ёњ Mn(OH)2(s) + H2
Ёя УЬФмШмгкЯЁЫсВЂЗХГіЧтЦјЁЃ
Ёя дкбѕЛЏМСДцдкЯТЃЌУЬФмгыШлШкЕФМюзїгУЩњГЩУЬЫсбЮЃК2Mn + 4KOH + 3O2 Ёњ 2K2MnO4 + 2H2O
Ёя УЬЛЙФмгыбѕЁЂТБЫиЕШЗЧН№ЪєзїгУЃЌЩњГЩЯргІЕФЛЏКЯЮяЁЃ
13.4.2 УЬЕФЛЏКЯЮя
УЬдзгЕФМлЕчзгЙЙаЭЮЊ3d54s2ЁЃУЬЕФзюИпбѕЛЏжЕЮЊ+7ЁЃУЬвВФмаЮГЩбѕЛЏжЕДг+6ЕН-2ЕФЛЏКЯЮяЁЃУЬЕФживЊЛЏКЯЮягаЃКИпУЬЫсМи(KMnO4)зЯКкЩЋОЇЬхЃЌУЬЫсМи(K2MnO4)АЕТЬЩЋОЇЬхЃЌЖўбѕЛЏУЬ(MnO2)КкЩЋЗлФЉЃЌСђЫсУЬ(MnSO4ЁЄ7H2O)ШтКьЩЋОЇЬхЃЌТШЛЏУЬ(MnCl2ЁЄ 4H2O)ШтКьЩЋОЇЬхЁЃ
вдШэУЬПѓЮЊдСЯЃЌПЩвджЦБИKMnO4,Mn3O4,MnOЕШЁЃжЦБИСїГЬЭМШчЯТЃК

13.4.3 УЬЕФЛЏКЯЮя
ЂБMn(Ђђ)ЕФаджЪ
Ёя Mn2+дкЫЎШмвКжаБШНЯЮШЖЈЃЌЫЎНтГЬЖШНЯаЁЁЃ
![]()
ЯђMn2+ ЕФШмвКжаМгШыOH-ЪБЃЌЯШЕУЕНАзЩЋЕФMn(OH)2ГСЕэЁЃ
Mn2+ + 2OH- Ёњ Mn(OH)2(s)
Mn(OH)2дкПеЦјжаКмПьБЛбѕЛЏГЩзиЩЋЕФMn3O4КЭ MnO2ЕФЫЎКЯЮя
![]()
Лђ ![]()
Ёя дкАБМюадЬѕМўЯТЃЌЯђMn2+ШмвКжаМгШы H2SШмвКЃЌЩњГЩШтКьЩЋЕФMnSГСЕэЁЃИУГСЕэПЩШмгкДзЫсжаЁЃ
Mn2+ + H2S + 2NH3 Ёњ MnS(s,ШтЩЋ) + 2NH4+
MnS + 2HAC Ёњ Mn2+ + H2S + 2AC-
Ёя Mn2+ЕФЛЙдадВюЃЌдкЫсадНщжЪжавЊЪЕЯж ![]() ЕФзЊЛЏашвЊВЩгУЧПбѕЛЏМСЁЃР§ШчЃКNaBiO3,PbO2,K2S2O8,H5IO6 ЕШЁЃ
ЕФзЊЛЏашвЊВЩгУЧПбѕЛЏМСЁЃР§ШчЃКNaBiO3,PbO2,K2S2O8,H5IO6 ЕШЁЃ
![]()
ЫЕУїЃК
Ђй ИУЗДгІГЃгУРДМјЖЈШмвКжаЮЂСПЕФMn2+ЁЃШєMn2+ДцдкЃЌдђШмвКгЩЮоЩЋ(Mn2+)БфЮЊзЯКьЩЋ( ![]() )ЁЃ
)ЁЃ
Ђк ЫсЛЏЪБВЛФмгУHClЃЌCl-ЛсЛЙд ![]() ЖјЪЙзЯЩЋСЂМДЭЪШЅЁЃ
ЖјЪЙзЯЩЋСЂМДЭЪШЅЁЃ
Ђл Mn2+ЕФХЈЖШКмЕЭЪБЃЌКмСщУєЁЃЕБMn2+Й§ЖрЪБвВЛсЪЙзЯЩЋСЂМДЯћЪЇЁЃ
![]()
ЂВ Mn(Ђє)ЕФаджЪ
Mn(Ђє)ЭЈГЃвдMnO2ЕФаЮЪНДцдкЁЃMnO2ЭЈГЃГЪЯжЧПбѕЛЏад ЁЃ
![]()
MnO2вВгавЛЖЈГЬЖШЕФЛЙдадЁЃ
MnO2 + 2MnO4Ѓ + 4OHЃ (ХЈ) Ёњ 3MnO42Ѓ + 2H2O
ЂГMn(Ђі)ЕФаджЪ
Mn(Ђі)вЛАувдK2MnO4ЕФаЮЪНДцдкЁЃK2MnO4ЪЧАЕТЬЩЋОЇЬхЃЌ ![]() дкpHЃО13.5ЕФЧПМюадШмвКжаВХФмДцдкЃЌдкЫЎШмвКЛђЫсадШмвКжавзЦчЛЏЃК
дкpHЃО13.5ЕФЧПМюадШмвКжаВХФмДцдкЃЌдкЫЎШмвКЛђЫсадШмвКжавзЦчЛЏЃК
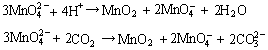
ЂДMn(Ђї)ЕФаджЪ
Mn(Ђї)ЭЈГЃвдKMnO4ЕФаЮЪНДцдкЁЃKMnO4гаЧПбѕЛЏадЃЌ ![]()
ПЩбѕЛЏЮяжЪ |
| I- | Cl- | H2S | Fe2+ | Sn2+ |
ВњЮя |
| I2 | Cl2 | SЛђ | Fe3+ | Sn4+ |
ШмвКЕФЫсЖШВЛЭЌЃЌ ![]() БЛЛЙдЕФВњЮявВВЛЭЌ
БЛЛЙдЕФВњЮявВВЛЭЌ
Ёя ЫсадЬѕМўЃК
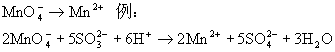
Ёя жаадЬѕМўЃК
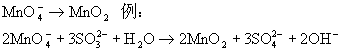
Ёя ХЈЧПМюадЬѕМўЬѕМўЃК
KMnO4 ШШЮШЖЈадВюЃЌЭЈГЃЪЂзАгкзиЩЋЦПжаЁЃР§ШчЃК
Ёя МћЙт(гіЫс)ЃК ![]()
Ёя ХЈМюЃК ![]()
Ёя МгШШЃК ![]()
Ёя
ЂЕУЬЕФгаЙиРызгКЭЛЏКЯЮяЕФаджЪаЁНс
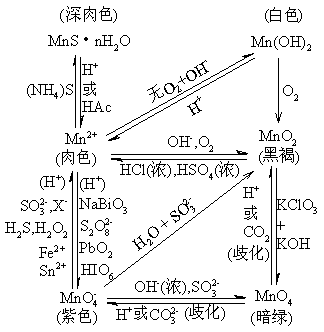
Ёь13.5ЁЁЬњ юм Фј
жмЦкЯЕЕкЂј зхдЊЫиАќРЈЃК
Ьњ(Fe) юм(Co) Фј(Ni) Ёћ ЬњЯЕ
юЩ (Ru) юю(Rh) юй (Pd) Ёћ ВЌЯЕ
яА (Os) вП(Ir) ВЌ(Pt) Ёћ ВЌЯЕ
ЬњЯЕдЊЫижаЃЌвдЬњЕФЗжВМзюЙуЁЃЬњдкЕиПЧжаЕФКЌСПОгЕкЫФЮЛЃЌдкН№ЪєжаНіДЮгкТСЁЃЬњЕФжївЊПѓЪЏгаГрЬњПѓ(Fe2O3)ЁЂДХЬњПѓ(Fe3O4)ЁЂЛЦЬњПѓ(FeS2)КЭСтЬњПѓ(FeCO3)ЕШЁЃюмКЭФјЕФГЃМћПѓЮяЪЧЛдюмПѓ(CoAsS)КЭФјЛЦЬњПѓ(NiSЁЄ FeS)ЁЃ
13.5.1 ЬњЁЂюмЁЂФјЕФЕЅжЪ
ЬњЁЂюмЁЂФјЖМЪЧвјАзЩЋН№ЪєЃЌФмБЛДХЬхЫљЮќв§ЃЌЪЧ ЬњДХадЮяжЪ ЁЃ
юмЁЂФјКЭДПЬњдкПеЦјжаЖМЪЧЮШЖЈЕФЃЌЕЋвЛАуЕФЬњвђКЌгадгжЪдкГБЪЊЕФПеЦјжаТ§Т§аЮГЩзиЩЋЕФЬњатFe2O3ЁЄxH2OЁЃ
ЬњЁЂюмЁЂФјЖМФмДгЯЁЫсжажУЛЛГіЧтЦјЁЃ Co,NiЕФЯргІЗДгІвЊТ§вЛаЉЁЃ
РфЕФЯѕЫсШмвКПЩЪЙЬњЁЂюмЁЂФјБфГЩЖлЬЌЁЃХЈСђЫсПЩЪЙЬњЖлЛЏЁЃЖлЬЌЕФЬњЁЂюмЁЂФјВЛдйШмгкЯргІЕФЫсжаЃЌЫљвдПЩвдгУЬњЙожќДцХЈСђЫс
дкМгШШЬѕМўЯТЃЌЬњЁЂюмЁЂФјФмгыаэЖрЗЧН№ЪєОчСвЗДгІЁЃ
13.5.2 ЬњЁЂюмЁЂФјЕФЛЏКЯЮя
| МлЕчзгЙЙаЭ | живЊбѕЛЏжЕ |
Fe | 3d64s2 | +3 +2 (+6) |
Co | 3d74s2 | +2 +3 (+5) |
Ni | 3d84s2 | +2 +3 (+4) |
зюИпбѕЛЏжЕВЛЕШгкзхађЪ§ЁЃетжївЊЪЧвђЮЊЫцзХдзгађЪ§ЕФдіМгЃЌдзгЕФгааЇКЫЕчКЩдіМгЃЌдіЧПСЫКЫЖд3dЕчзгЕФЪјИПзїгУ
ЬњЁЂюмЁЂФјЕФИпбѕЛЏЬЌЛЏКЯЮябѕЛЏадНЯЧПЃЌДѓЖМвдКЌбѕЫсбЮЛђХфбЮЕФаЮЪНДцдкЃЌР§ШчЃКNa2FeO4,K3CoO4,K2NiF6ЕШЃЌетРрЛЏКЯЮядкЫЎШмвКжаЖММЋВЛЮШЖЈЁЃ
Fe3+,Co3+,Ni3+ЕФбѕЛЏадАДFe3+ЃМCo3+ЃМNi3+ЫГађдіЧПЃЌвђДЫЫќУЧТБЛЏЮяЮШЖЈДцдкзДПіМћЯТБэЃК
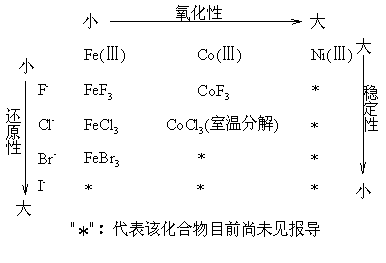
FeCl3гаУїЯдЕФЙВМладЃЌвзГБНтЁЃЫќЕФеєЦјжаКЌгаЫЋОлЗжзг Fe2Cl6ЃЌЦфНсЙЙЮЊЃК
ТШЛЏюмCoCl2ЁЄ6H2OдкЪмШШЭбЫЎЙ§ГЬжаЃЌАщЫцзХбеЩЋЕФБфЛЏЃК
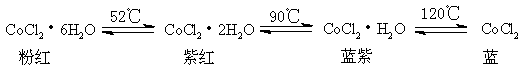
РћгУCoCl2ЁЄ6H2OЕФетвЛЬиаджИЪОГіЕФЮќЫЎГЬЖШЁЃ
ЬњЁЂюмЁЂФјЕФЦфЫќЛЏКЯЮяОпгабЮРрЕФвЛАуаджЪЁЃ
13.5.3 ЫЎШмвКжаЬњЁЂюмЁЂФјЕФРызгМАЦфЗДгІ
ЂБFe(Ђђ )ЁЂFe(Ђѓ)ЁЂ Co(Ђђ )ЁЂNi(Ђђ)ЕФЫЎНтад
Fe(Ђђ)ЁЂFe(Ђѓ )дкЫЎШмвКжаЗжБ№вд[Fe(H2O)6]3+(ЕзЯЩЋ)КЭ[Fe(H2O)6]2+(ЕТЬЩЋ)ЕФаЮЪНДцдкЁЃюмКЭФјЕФбЮРрдкЫЎШмвКжажївЊга[Co(H2O)6]3+(ЗлКьЩЋ) КЭ[Ni(H2O)6]3+(ТЬЩЋ) ЁЃ
гЩгкFe3+БШFe2+ЕФЕчКЩЖрЃЌвђЖјFe3+БШFe2+ШнвзЗЂЩњЫЎНтЁЃ
![]()
![]()
Co2+КЭNi2+НіЗЂЩњЮЂШѕЕФЫЎНтЁЃ
![]()
![]()
гЩгкFe3+ЫЎНтГЬЖШДѓЃЌ[Fe(H2O)6]3+НіФмДцдкгкЫсадНЯЧПЕФШмвКжаЃЌЯЁЪЭШмвКЛђдіДѓШмвКЕФpHжЕ ,ЖМЛсгаНКзДЮяFeO(OH)ГСЕэГіРДЃЌЪЙЛьзЧЕФЫЎБфЧхЁЃЫљвдFeCl3ГЃгУзїОЛЫЎМСЁЃ
ЂВГСЕэЗДгІ
ЯђFe2+,Fe3+,Co2+,Ni2+ШмвКжаМгШыЧПМюЃЌЗжБ№ЩњГЩЯргІЕФЧтбѕЛЏЮяЁЃ
Fe2+ + 2OH- Ёњ Fe(OH)2(s,АзЩЋ)
Fe3+ + 3OH- Ёњ Fe(OH)3(s,КьзиЩЋ)
Co2+ + 2OH- Ёњ Co(OH)2(s,ЗлКьЩЋ)
Ni2+ + 2OH- Ёњ Ni(OH)2(s,ЦЛЙћТЬЩЋ)
Ёя Fe(OH)2бИЫйБЛПеЦјжаЕФбѕбѕЛЏЃЌгЩАзЩЋЁњЛвТЬЩЋЁњзиКжЩЋЃЌзюжеЩњГЩFe(OH)3 ЁЃ
Ёя ЪЊЕФ Co(OH)2вВФмБЛПеЦјжаЕФбѕЛКТ§ЕибѕЛЏГЩАЕзиЩЋЕФCo2O3ЁЄxH2O(Co(OH)3)ЃК
4Co(OH)2 + O2 + 2(x-2)H2O Ёњ 2Co2O3ЁЄxH2O
Ёя Ni(OH)2ашвЊдкХЈМюШмвКжагУНЯЧПЕФбѕЛЏМС(ШчфхЫЎ)ВХФмАбЫќбѕЛЏГЩКкЩЋЕФNiO(OH)(Ni(OH)3)ЁЃ
2Ni(OH)2 + Br2 + 2OH - Ёњ 2NiO(OH) + 2Br - + 2 H2O
Ёя Co(OH)2дкЭЌбљЬѕМўЯТвВПЩБЛбѕЛЏЮЊCo(OH)3ЁЃ
2Co(OH)2 + Br2 + 2OH- Ёњ 2Co(OH)3 + 2Br-
Ёя гЩгкCo3+,Ni3+ОпгаКмЧПЕФбѕЛЏадЃЌдкЫЎШмвКжаКмФбга [Co(H2O)6]3+КЭ[Ni(H2O)6]3+ДцдкЁЃЫљвдCo2O3,NiO(OH)гыЫсзїгУЪБЃЌЗЂЩњбѕЛЏЛЙдЗДгІЃК
2M(OH)3 + 6HCl(ХЈ) Ёњ 2MCl2 + Cl2 + 6H2O (M=Co,Ni)
дкАБМюадЬѕМўЯТЃЌЯђFe2+,Co2+,Ni2+ШмвКжаЭЈШыH2S,ФмЙЛЩњГЩMSГСЕэЁЃ
FeS,CoS,NiSЪЧЯЁЫсШмадЃЌМДдкЯЁЫсЬѕМўЯТЃЌЯђКЌга Fe2+,Co2+,Ni2+РызгЕФШмвКжаЭЈШыH2SЪЧВЛЛсВњЩњСђЛЏЮяГСЕэЕФЁЃЕЋЕБMSдкАБМюадЬѕМўЯТаЮГЩКѓЃЌCoS,NiSгЩгкЗЂЩњОЇаЭзЊБфЖјВЛдйШмгкЫсЁЃ
ЂГбѕЛЏЛЙдЗДгІ
ЬњЕФдЊЫиЕчЪЦЭМЃК
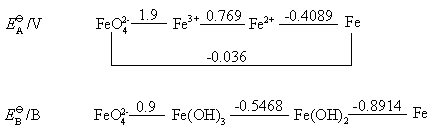
Ёя дкХЈМюШмвКжаЃЌгУNaClOПЩвдАбFe(OH)3бѕЛЏГЩзЯКьЩЋЕФFeO42-ЃК
2Fe(OH)3 + 3ClO- + 4OH- Ёњ 2FeO42- + 3Cl- + 5H2O
Ёя ЫсадШмвКжаЃЌFe3+ЪЧжаЧПбѕЛЏМСЃЌФмАбI-,H2S,Fe,CuЕШбѕЛЏЃК
2Fe3+ + H2S Ёњ 2Fe2+ + S + 2H+
2Fe3+ + Fe Ёњ 3Fe2+
2Fe3+ + Cu Ёњ 2Fe2+ + Cu2+
ЙЄвЕЩЯГЃгУFeCl3ЕФШмвКдкЬњжЦЦЗЩЯПЬЪДзжбљЃЌЛђдкЭАхЩЯжЦдьгЁЫЂЕчТЗЁЃFeCl3ШмвКвВНазіРУАхМСЁЃ
Ёя дкЫсадШмвКжаЃЌFe2+гавЛЖЈЕФЛЙдад(ЯдШЛFe2+ЕФЛЙдадБШFe(OH)2Шѕ) (ЕуЛїЭМЦЌЪЕбщбнЪО)ЁЃР§ШчЃКПеЦјжаЕФбѕФмАбFe2+бѕЛЏЮЊFe3+ЃК
4Fe2+ + O2 + 4H+ Ёњ 4Fe3+ + 2H2O
ЫљвдБЃДц Fe2+ШмвКЪБЃЌгІМгШыFeЁЃ
ЂДХфКЯЗДгІ
Ёя АБХфКЯЮя
Fe2+,Fe3+гыNH3ВЛаЮГЩХфКЯЮяЃК
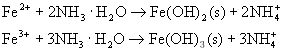
дкCo2+ЃЌNi2+ЕФШмвКжаМгШыАБЫЎЃЌЯШЩњГЩМюЪНбЮГСЕэЃЌЕБАБЫЎЙ§СПЪБЃЌаЮГЩАБХфКЯЮяЁЃ
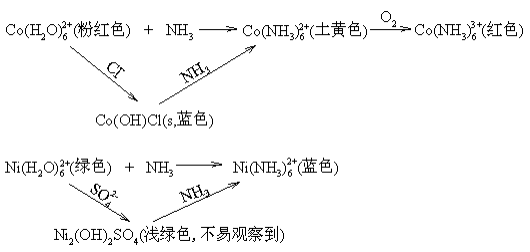
[Co(NH3)6]2+ОпгаНЯЧПЕФЛЙдад ,взБЛПеЦјжаЕФбѕбѕЛЏЮЊ[Co(NH3)6]3+:
Ёя СђЧшХфКЯЮя
МјЖЈCo2+
дкМјЖЈCo2+ЕФЪЕбщжаЃЌгУЙЬЬхЕФKSCNЛђNH4SCNв§ШыSCN-ИљЁЃ
![]()
Ёя ЧшЕФХфКЯЮя
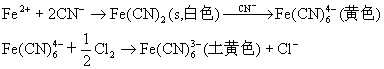
ЖдгІЕФбЮРргаЃКСљЧшКЯЬњ(Ђђ )ЫсМиЃКK4[Fe(CN)6]ЫзГЦЛЦбЊбЮЃЌГЪЛЦЩЋЁЃ
СљЧшКЯЬњ(Ђѓ)ЫсМиЃКK3[Fe(CN)6]ЫзГЦГрбЊбЮЃЌОЇЬхГЪКьЩЋЁЃ
![]() ,
, ![]() дкМюадЬѕМўЯТЖМВЛЮШЖЈЃЌЗжНтЮЊЯргІЕФЧтбѕЛЏЮяЃК
дкМюадЬѕМўЯТЖМВЛЮШЖЈЃЌЗжНтЮЊЯргІЕФЧтбѕЛЏЮяЃК
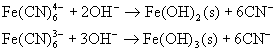
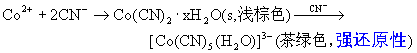

ЂЕгаЙиРызгЕФМјЖЈ
Ёя ЬњРызгЕФМјЖЈ
Fe2+ЕФМјЖЈЃКдкЫсадЬѕМўЯТМгШыK3[Fe(CN)6]ШмвК ЁЃ
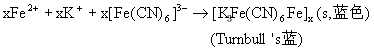
Fe3+ЕФМјЖЈ:Г§СЫПЩгУKSCNЭтЃЌЛЙПЩгУ K4[Fe(CN)6] ЁЃ
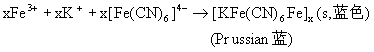
ЪЕбщвбжЄУїЃЌPrussianРЖКЭTurnbull'sРЖЕФзщГЩЖМЪЧ[KFeЂѓ(CN)6FeЂђ]xЁЃ
Ёя Cu2+ЕФМјЖЈЃКдкШѕЫсадЬѕМўЯТЃЌгУK4[Fe(CN)6]ШмвКЁЃ
2Cu2+ + [Fe(CN)6]4- Ёњ Cu2[Fe(CN)6](s,КьзиЩЋ)
Ёя S2-ЕФМјЖЈЃКРћгУ[Fe(CN)6]4-гыЯѕЫсзїгУКѓЩњГЩЕФ[Fe(CN)5NO]2-МјЖЈS2-ЕФДцдкЃК
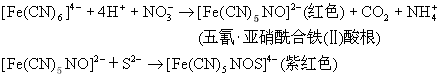
Ёя K+ЕФМјЖЈЃКАбNa3[Co(NO2)6]ШмвКМгЕНКЌга K+ЕФШмвКжаЃЌдђЮіГіФбШмгкЫЎЕФЛЦЩЋОЇЬхK2Na[Co(NO2)6]ЃК
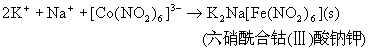
Ёя Ni2+ЕФМјЖЈЃКдкШѕМюадЬѕМўЯТМгШыЖЁЖўыПЩњГЩФбШмгкЫЎЕФЯЪКьЩЋђќКЯЮяГСЕэЖўЖЁЖўыПКЯФј(Ђђ ) ЃК
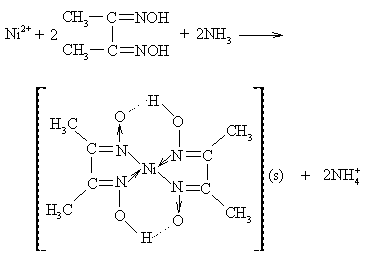
ЖўЖЁЖўыПКЯФјМђаДзїЃК Ni(DMG)2ЁЃ
ЂЖживЊЗДгІаЁНс
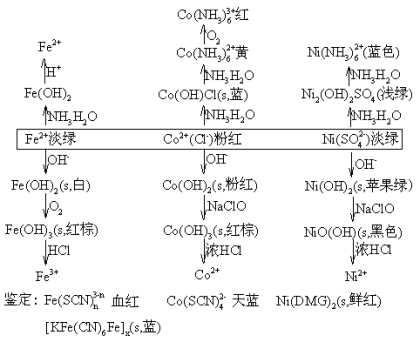
ЕкЪЎЫФеТ Й§ЖЩдЊЫи(Жў)
БОеТЮвУЧжївЊСЫНтЭзхдЊЫиЕФЭЈадЁЃСЫНтЭЕФбѕЛЏЮяЁЂЧтЛЏЮяЁЂживЊЭбЮЕФаджЪЃЌCu(Ђё)КЭCu(Ђђ)ЕФЯрЛЅзЊЛЏЃЌЭЕФХфКЯЮяЁЃ ЪьЯЄвјЕФбѕЛЏЮяЁЂЧтбѕЛЏЮяЕФаджЪЃЌвјЕФживЊХфКЯЮяЁЃСЫНтаПзхдЊЫиЕФЭЈадЁЃеЦЮеЧтбѕЛЏаПЕФаджЪЁЂЫЎШмвКжаZn2+ЕФживЊЗДгІЁЂаПЕФживЊХфКЯЮяЁЃЪьЯЄаПЁЂягЁЂЙЏЕФбѕЛЏЮяЃЌягЁЂЙЏЕФЧтбѕЛЏЮяЕФаджЪЃЌHg(Ђё)КЭHg(Ђђ)МфЕФЯрЛЅзЊЛЏЃЌягЁЂЙЏЕФХфКЯЮяЁЃ
Ёь14.1ЁЁЭзхдЊЫи
14.1.1 ЭзхдЊЫиМђНщ
жмЦкЯЕЕкЂёBдЊЫиЃЌАќРЈЭ(Cu)ЁЂвј(Ag)ЁЂН№(Au)3жждЊЫиЃЌЭЈГЃГЦЮЊЭзхдЊЫиЁЃМлЕчзгЙЙаЭЮЊ(n-1)d10ns1ЁЃ
дкздШЛНчжаЃЌЭзхдЊЫиГ§СЫвдПѓЮяаЮЪНДцдкЭтЃЌЛЙвдЕЅжЪаЮЪНДцдкЁЃГЃМћЕФПѓЮягаЛдЭПѓ(Cu2S)ЁЂПзШИЪЏ[Cu2(OH)2CO4]ЁЂЛдвјПѓ(Ag2S)ЁЂэкН№Пѓ(AuTe2)ЕШЁЃ
14.1.2 ЭзхдЊЫиЕФЕЅжЪ
1.ЮяРэаджЪ
Ёя ЭЁЂвјЁЂН№ЖМгаЬиеїбеЩЋЃКCu(зЯКь)ЁЂAg(Аз)ЁЂAu(ЛЦ)ЁЃ
Ёя ЭЁЂвјЁЂН№ЕФШлЗаЕуВЛЬЋИпЁЃ
Ёя ЫќУЧЕФЕМЕчадЁЂЕМШШадЁЂбгеЙадЬиБ№ЭЛГіЁЃЫќУЧЕФЕМЕчадЫГађЮЊЃКAgЃОCuЃОAuЁЃгЩгкЭЕФМлИёНЯЕЭЃЌЫљвдЃЌЭдкЕчЦїЙЄвЕЩЯЕУЕНСЫЙуЗКЕФгІгУЁЃ
ЂВЛЏбЇаджЪ
ЭЁЂвјЁЂН№ЕФЛЏбЇЛюЦУадНЯВюЃЌдкЪвЮТЯТПДВЛГіЫќУЧгыбѕЛђЫЎзїгУЁЃдкКЌгаCO2ЕФГБЪЊПеЦјжаЃЌЭЕФБэУцЛсж№НЅУЩЩЯТЬЩЋЕФЭат(ЭТЬЁЊЬМЫсєЧЭCu2(OH)2CO3)ЁЃ
2Cu + O2 + H2O + CO2 Ёњ Cu2(OH)2CO3
дкМгШШЬѕМўЯТЃЌЭгыбѕЛЏКЯГЩCuO,ЖјвјЁЂН№ВЛЗЂЩњБфЛЏЁЃДЫЫљЮНЁАецН№ВЛХТЛ№СЖЁБЃЁ
зЂвтЃКЕБГСЕэМСЛђХфКЯМСДцдкЪБЃЌЭЁЂвјЁЂН№вВПЩгыбѕЗЂЩњзїгУЃК
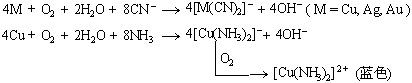
14.1.3 ЭзхдЊЫиЕФЛЏКЯЮя
ЂБЭЕФЛЏКЯЮя
ЭЕФГЃМћЛЏКЯЮяЕФбѕЛЏжЕЮЊ+1КЭ+2ЁЃCu(Ђё)ЮЊd10ЙЙаЭЃЌУЛгаdЁЊdдОЧЈЃЌCu(Ђё)ЕФЛЏКЯЮявЛАуЪЧАзЩЋЛђЮоЩЋЕФЁЃCu(Ђђ)ЮЊd9ЙЙаЭЃЌЫќУЧЕФЛЏКЯЮяжаГЃвђCu2+ЗЂЩњdЁЊdдОЧЈЖјГЪЯжбеЩЋЁЃ
Ёя вЛАуЫЕРДЃЌдкИпЮТЁЂЙЬЬЌЪБЃЌCu(Ђё)ЕФЛЏКЯЮяБШCu(Ђђ)ЕФЛЏКЯЮяЮШЖЈЃЌР§ШчЃК
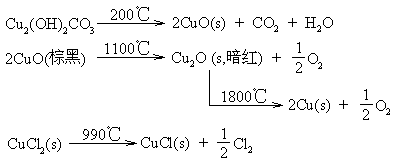
Ёя дкЫЎШмвКжаЃЌCu(Ђё)взБЛбѕЛЏЮЊCu(Ђђ)ЃЌЫЎШмвКжаCu(Ђђ)ЕФЛЏКЯЮяНЯЮШЖЈЁЃ
Ёя Cu(Ђё)ЕФЛЏКЯЮяЖМФбШмгкЫЎЃЌГЃМћЕФCu(Ђё)ЛЏКЯЮядкЫЎжаЕФШмНтЖШЫГађЮЊЃК
CuClЃОCuBrЃОCuIЃОCuSCNЃОCuCNЃОCu2S
Cu(Ђђ)ЕФЛЏКЯЮявзШмгкЫЎЕФНЯЖрЁЃ
ГЃМћЕФЮхЫЎСђЫсЭ(CuSO4ЁЄ5H2O)ЫзГЦЕЈЗЏЃЌОЇЬхГЪРЖЩЋЃЌЭъШЋЭбЫЎКѓБфЮЊАзЩЋЗлФЉCuSO4
![]()
ЮоЫЎCuSO4взЮќЫЎЃЌЮќЫЎКѓГЪРЖЩЋЃЌГЃБЛгУРДМјЖЈвКЬЌгаЛњЮяжаЕФЮЂСПЫЎЁЃЙЄвЕЩЯГЃгУСђЫсЭзїЮЊЕчНтЭЕФдСЯЁЃдкХЉвЕЩЯЃЌгУЫќгыЪЏЛвШщЕФЛьКЯвКРДЯћУ№ЙћЪїЩЯЕФКІГцЁЃ
ЂВвјЁЂН№ЕФЛЏКЯЮя
дквјЕФЛЏКЯЮяжаЃЌAg(Ђё)ЕФЛЏКЯЮязюЮШЖЈЃЌЖјН№дђвдAu(Ђѓ)ЕФЛЏКЯЮяНЯЮЊГЃМћЃЌЕЋдкЫЎШмвКжаЖрвдХфКЯЮяаЮЪНДцдкЁЃ
вјЕФЛЏКЯЮяОпгавдЯТЬиЕуЃК
Ёя ФбШмЕФЖрЁЃ
взШмЃКAgNO3, AgF, AgClO4
ФбШмЃКAgCl, AgBr, AgI, AgCN, AgSCN, Ag2S, Ag2CO3, Ag2CrO4ЕШЁЃ
Ёя ШШЮШЖЈадВю(МћЙтЃЌЪмШШвзЗжНт)ЁЃ
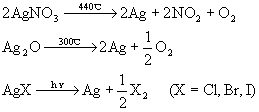
Ёя габеЩЋЁЃ
AgCl AgBr AgI Ag2O Ag2CrO4 Ag2S
Аз ЧГЛЦ ЛЦ Кж зЉКь Кк
14.1.4 ЫЎШмвКжаЭзхдЊЫиЕФРызгМАЦфЗДгІ
ЂБCu(Ђђ)ЁЂCu(Ђё)РызгЕФЗДгІ
Ёя [Cu(H2O)6]2+ГЪРЖЩЋЃЌЫЎНтГЬЖШаЁЁЃ
![]()
OH-(ЪЪСП) OH-(Й§СП)
Cu2+ ![]() Cu (OH)2(ЧГРЖ)
Cu (OH)2(ЧГРЖ) ![]() Cu (OH)4
Cu (OH)4
| |
|
|
![]() 80~90Ёц
80~90Ёц
CuO
![]() гыЦЯЬбЬЧдкМгШШЬѕМўЯТЗДгІЃЌгаАЕКьЩЋЕФCu2OГСЕэЮіГіЃК
гыЦЯЬбЬЧдкМгШШЬѕМўЯТЗДгІЃЌгаАЕКьЩЋЕФCu2OГСЕэЮіГіЃК
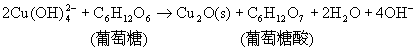
етвЛЗДгІдкгаЛњЛЏбЇжагУРДМьбщФГаЉЬЧЕФДцдкЁЃ
Ёя [Cu(H2O)6]+ЪЧЮоЩЋЕФЃЌЫЎШмвКжаКмВЛЮШЖЈЃЌШнвзЦчЛЏЮЊCu2+КЭCuЃК
![]()
Р§ШчЃК Cu2O + H2SO4 Ёњ CuSO4 + Cu + H2O
ЫљвддкЫЎШмвКжаCu(Ђђ)БШCu(Ђё)ЮШЖЈЁЃ
Ёя гаХфКЯМСЁЂГСЕэМСДцдкЪБЃЌCu(Ђё)ЕФЮШЖЈадЬсИпЁЃ
Cu2O + 2HCl Ёњ 2CuCl(s) + H2O
Cu2+ + 4Cl- + Cu Ёњ 2[CuCl2]-(ФрЛЦЫи)
ЫљвдЃЌГЃРћгУCuSO4ЛђCuCl2ШмвКгыХЈбЮЫсКЭЭаМЛьКЯЃЌдкМгШШЬѕМўЯТжЦШЁ[CuCl2]-ШмвКЁЃ
Ёя Cu2+ОпгавЛЖЈЕФбѕЛЏадЃК
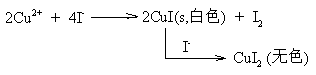
ЫМПМЃКШчКЮМјЖЈШмвКжаЪЧЗёДцдкCu2+ЃП
Ёя Cu(Ђё)ЕФХфКЯЮяЖрЮЛ2ХфЮЛЕФЃЌЦфЮШЖЈадЫГађЮЊЃК
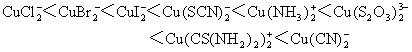
Cu(Ђђ)ЕФХфКЯЮяЖрЮЛ4ХфЮЛЕФЃЌР§ШчЃК
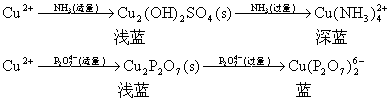
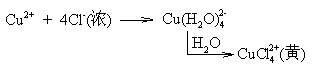
CuSO4ЁЄ5H2OвВЪЧХфКЯЮяЃЌМД[Cu(H2O)4]SO4ЁЄH2O
ЂВAg(Ђё)РызгЕФЗДгІ
Ёя вЛАуШЯЮЊЫЎКЯвјРызгЕФЛЏбЇЪНЪЧ[Ag(H2O)4]+,ЫќдкЫЎжаМИКѕВЛЫЎНтЁЃAgNO3ЕФЫЎШмвКГЪжаадЁЃдкAg+жаМгШыNaOHШмвКЃЌвђЮЊAgOHМЋВЛЮШЖЈЃЌЮіГіЕФГСЕэЪЧAg2OЃК
2Ag+ + 2OH- Ёњ Ag2O(s) + H2O
Ёя Ag(Ђё)ЕФаэЖрЛЏКЯЮяЖМЪЧФбШмгкЫЎЕФЃЌдкAg+ЕФШмвКжаМгШыХфЮЛМСЪБЃЌГЃГЃЯШЕФЩњГЩФбШмЛЏКЯЮяЃЌЕБХфЮЛМСЙ§СПЪБЃЌФбШмЛЏКЯЮяШмНтЩњГЩХфРызгЁЃР§ШчЃК
![]()
КЌга[Ag(NH3)2]+ЕФШмвКФмАбШЉЛђФГаЉЬЧбѕЛЏЃЌБОЩэБЛЛЙдЮЊЕЅжЪвјЁЃ
2[Ag(NH3)2]+ + HCHO + 3OH- Ёњ 2Ag(s) + HCOO- + 4NH3 + 2H2O
етРрЗДгІвВНазівјОЕЗДгІЃЌЙЄвЕЩЯРћгУетРрЗДгІРДжЦзїОЕзгЛђдкХЏЫЎЦПЕФМаВуФкЖЦвјЁЃ
гжШчЃКAg+гы ![]() Лђ
Лђ ![]() ЗДгІЖМЩњГЩAg2CrO4,Ag2CrO4ПЩШмгкзуСПЕФАБЫЎжаЃК
ЗДгІЖМЩњГЩAg2CrO4,Ag2CrO4ПЩШмгкзуСПЕФАБЫЎжаЃК
![]()
ИУаджЪгУгкBa2+КЭAg+ЕФЗжРыЁЃ(ЧызЂвтШчКЮОпЬхВйзїЃП)
Ag+гыЩйСПNa2S2O3ШмвКЗДгІЩњГЩAg2S2O3АзЩЋГСЕэЃЌЗХжУвЛЖЮЪБМфжЎКѓЃЌГСЕэгЩАзЩЋзЊБфЮЊЛЦЩЋЁЂзиЩЋзюКѓЮЊКкЩЋAg2SЁЃ
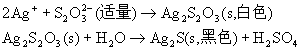
ЕБNa2S2O3Й§СПЪБЃЌAg2S2O3ШмНтЃЌЩњГЩХфРызг[Ag(S2O3)2]3-ЃК
![]()
Ag2SЕФШмНтЖШКмаЁЃЌФбвдНшХфЮЛЗДгІЪЙЫќШмНтЃЌвЛАуВЩгУHNO3ЕФбѕЛЏадРДЪЕЯжAg2SШмНтЃК
![]()
ИХРЈвдЩЯЫЎШмвКжаAg+РызгЕФаджЪЃЌПЩЭЈЙ§вдЯТ"Ag+ађ"зїМђЕЅаЁНсЃК
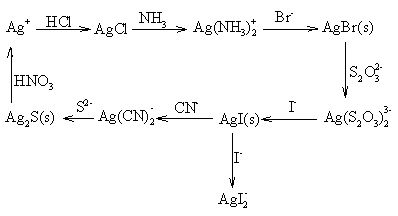
Ёя Ag+ЕФМјЖЈЁЃ
дкAg+ЕФМјЖЈЪЕбщжаЃЌМгШыHClгаАзЩЋГСЕэЩњГЩНіЫЕУїПЩФмгаAg+ЃЌБиаызїНјвЛВНЕФМјЖЈЁЃМДМгШыNH3ЁЄH2OГСЕэШмНтЃЌНЋИУШмвКЫсЛЏЃЌгаАзЩЋГСЕэВњЩњЛђдкШмвКжаМгШыKIЃЌгаЛЦЩЋГСЕэВњЩњЃЌВХФмжЄУїдЪМШмвКжаШЗЪЕДцдкAg+ЁЃЭМЪОШчЯТЃК
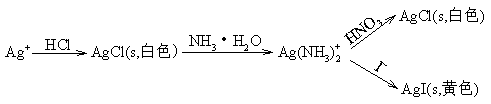
жївЊЗДгІгаЃК
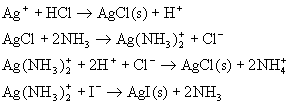
Ёь14.2ЁЁаПзхдЊЫи
14.2.1 аПзхдЊЫиМђНщ
жмЦкЯЕЕкЂђBдЊЫиЃЌАќРЈаП(Zn)ЁЂяг(Cd)ЁЂЙЏ(Hg)3жждЊЫиЃЌЭЈГЃГЦЮЊаПзхдЊЫиЁЃЫќУЧЪЧгыpЧјдЊЫиЯрСкЕФdЧјдЊЫиЃЌОпгагыdЧјдЊЫиЯрЫЦЕФаджЪЃЌШчвзгкаЮГЩХфКЯЮяЕШЁЃдкФГаЉаджЪЩЯЫќУЧгжгыЕкЫФЁЂЮхЁЂСљжмЦкЕФpЧјН№ЪєдЊЫигааЉЯрЫЦЃЌШчШлЕуЕЭЃЌЫЎКЯРызгЖМЮоЩЋЕШЁЃаПзхдЊЫивЛАувдПѓЮяаЮЪНДцдкЃЌР§ШчЩСаППѓ(ZnS)ЁЂЩА(HgS)ЕШЁЃ
14.2.2 аПзхдЊЫиЕФЕЅжЪ
1.ЮяРэаджЪ
Ёя аПЁЂягЁЂЙЏЖМЪЧвјАзЩЋН№Ъє(аПТдДјРЖЩЋ)ЁЃ
Ёя ЫќУЧЖМЪЧЕЭШлЕуН№ЪєЃЌЬиБ№ЪЧЙЏЃЌЫќЪЧЪвЮТЯТЮЈвЛЕФвКЬЌН№ЪєЁЃ
Ёя взаЮГЩКЯН№ЃЌР§ШчЛЦЭCuЁЊZnЃЛЙЏЦыNaЁЊHgЃЌAuЁЊHgЃЌAgЁЊHgЁЃ
дк0ЁцЁЋ200ЁцжЎМфЃЌЙЏЕФХђеЭЯЕЪ§ЫцзХЮТЖШЩ§ИпЖјОљдШЕиИФБфЃЌВЂЧвВЛШѓЪЊВЃСЇЃЌдкжЦдьЮТЖШМЦЪБГЃРћгУЙЏЕФетвЛаджЪЁЃСэЭтвВгУЙЏЬюГфдкЦјбЙМЦжаЁЃФЦЙЏЦыдкгаЛњКЯГЩжагУзїЮТКЭЕФЛЙдМСЃЌН№ЙЏЦыЁЂвјЙЏЦыгУгкЬсДПЙѓН№ЪєЁЃ
ЂВЛЏбЇаджЪ
Ёя гыбѕЕФзїгУЁЃ
аПЁЂягЁЂЙЏдкИЩдяЕФПеЦјжаЖМЪЧЮШЖЈЕФЁЃдкгаCO2ДцдкЕФГБЪЊПеЦјжаЃЌаПЕФБэУцГЃЩњГЩвЛВуМюЪНЬМЫсбЮЕФБЁФЄЃЌБЃЛЄаПВЛБЛМЬајбѕЛЏЁЃ
4Zn + 2O2 + CO2 + 3H2 Ёњ ZnCO3ЁЄ3Zn(OH)2(МюЪНЬМЫсаП)
дкПеЦјжаМгШШаПЁЂягЁЂЙЏЖМФмаЮГЩЯргІЕФбѕЛЏЮяЁЃ
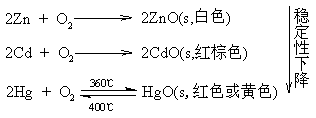
Ёя гыСђЕШЗЧН№ЪєЕФзїгУЁЃ
аПЁЂягЁЂЙЏОљФмгыСђЗлзїгУЃЌЩњГЩЯргІЕФСђЛЏЮяЁЃЬиБ№ЪЧЙЏЃЌдкЪвЮТЯТОЭПЩвдгыСђЗлзїгУЃЌЩњГЩHgSЁЃЫљвдПЩвдАбСђЗлШідкгаЙЏЕФЕиЗНЃЌЗРжЙгаЖОЕФЙЏеєЦјНјШыПеЦјжаЁЃШєПеЦјжавбгаЙЏеєЦјЃЌПЩвдАбЕтЩ§ЛЊЮЊЦјЬхЃЌЪЙЙЏеєЦјгыЕтеєЦјЯргіЃЌЩњГЩHgI2ЃЌвдГіШЅПеЦјжаЕФЙБеєЦјЁЃ
Ёя гыЫсЗДгІЁЃ
аПЁЂягФмДгЯЁЫсжажУЛЛГіЧтЦјЁЃЙЏФмгыЯѕЫсЗДгІЖјШмНтЁЃ
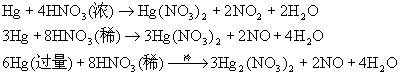
Ёя аПгыOH-,NH3ЗДгІЁЃ
аППЩвдДгМюШмвКжажУЛЛГіЧтЦјЁЃ
![]()
14.2.3 аПзхдЊЫиЕФЛЏКЯЮя
аПЁЂягЁЂЙЏЕФМлЕчзгЙЙаЭЮЊ(n-1)d10ns2ЁЃЫљвдаПКЭЙЏЭЈГЃаЮГЩбѕЛЏжЕЮЊ+2ЕФЛЏКЯЮяЃЌЙЏГ§СЫаЮГЩбѕЛЏжЕЮЊ+2ЕФЛЏКЯЮяЭтЃЌЛЙгабѕЛЏжЕЮЊ+1( ![]() )ЕФЛЏКЯЮяЁЃаПКЭягЕФЛЏКЯЮяОпгаДѓЖрЪ§ЮоЛњбЮЕФвЛАуЭЈадЃЌДЫДІОЭжиЕуЬжТлЙЏЕФЛЏКЯЮяЁЃ
)ЕФЛЏКЯЮяЁЃаПКЭягЕФЛЏКЯЮяОпгаДѓЖрЪ§ЮоЛњбЮЕФвЛАуЭЈадЃЌДЫДІОЭжиЕуЬжТлЙЏЕФЛЏКЯЮяЁЃ
Ёя ЙЏЕФТБЛЏЮяЁЃ
ТШЛЏЙЏ(HgCl2вВГЦЩ§ЙЏ)ЃЌЪЧжБЯпаЭЙВМлЗжзгЃЌвзЩ§ЛЊЃЌЮЂШмгкЫЎЃЌдкЫЎШмвКжажївЊвдЗжзгаЮЪНДцдкЃЌОчЖОЁЃ
ТШЛЏбЧЙЏ(Hg2Cl2вВГЦИЪЙЏ)ЃЌвВЪЧжБЯпаЭЗжзг(ClЁЊHgЁЊHgЁЊCl)ЃЌгаЬ№ЮЖЃЌФбШмгкЫЎЃЌЮоЖОЁЃ
HgBr2ЃЌHgI2ЭЌHgCl2ЃЌЕЋHgF2ЮЊРызгаЭЛЏКЯЮяЁЃ
Ёя ЙЏЕФЯѕЫсбЮЁЃ
ЯѕЫсЙЏHg(NO3)2КЭЯѕЫсбЧЙЏHg2(NO3)2ЪЧРызгаЭЛЏКЯЮяЃЌвзШмгкЫЎЁЃHg(NO3)2ПЩгУHgOЛђHgЯѕЫсзїгУжЦШЁЃК
HgO + 2HNO3 Ёњ Hg(NO3)2 + H2O
Hg + 4HNO3(ХЈ) Ёњ Hg(NO3)2 + 2NO2 + 2H2O
ЯѕЫсЙЏHg(NO3)2гыHgзїгУПЩжЦШЁHg2(NO3)2ЃК
Hg(NO3)2 + Hg Ёњ Hg2(NO3)2
гЩЕчЪЦЭМЗжЮіЃК
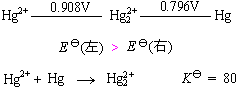
![]() ВЛЗЂЩњЦчЛЏЁЃ
ВЛЗЂЩњЦчЛЏЁЃ
14.2.4 ЫЎШмвКжааПзхдЊЫиЕФРызгМАЦфаджЪ
1.Zn2+,Cd2+РызгЕФЗДгІ
Ёя ЫЎНтЗДгІЃК
[Zn(H2O)6]2+КЭ[Cd(H2O)6]2+ЕФЫЎНтЧїЪЦЖМНЯШѕЃК
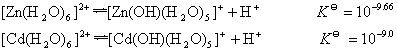
Ёя ГСЕэЗДгІЃК
дкZn2+,Cd2+ЕФШмвКжаМгШыЧПМюЃЌЖМЩњГЩЧтбѕЛЏЮяЃЌЕЋZn(OH)2ЪЧСНадЃЌЖјCd(OH)2ЪЧМюадЕФЁЃ
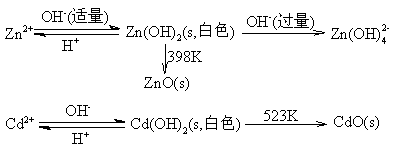
дкZn2+,Cd2+ЕФШмвКжаЗжБ№ЭЈШыH2SЪБЃЌЖМЛсгаСђЛЏЮяДгШмвКжаГСЕэГіРДЃК
Zn2+ + H2S Ёњ ZnS(s,АзЩЋ) + 2H+
Cd2+ + H2S Ёњ CdS(s,ЛЦЩЋ) + 2H+
зЂвтЃКZn2+жЛгадкАБМюадЬѕМўЯТВХФмГСЕэЭъШЋЁЃCdSЕФЛЦЩЋПЩзїЮЊМјЖЈCd2+ЕФЬиеїбеЩЋЁЃ
дкZnSO4ШмвКжаМгШыBaSЪБЃЌЩњГЩZnSКЭBaSO4ЕФЛьКЯГСЕэЮяЃК
![]()
ДЫГСЕэНазіаПБЕАзЃЌЫзГЦСЂЕТЗлЃЌЪЧвЛжжНЯКУЕФАзЩЋбеСЯЃЌУЛгаЖОадЃЌдкПеЦјжаБШНЯЮШЖЈЁЃ
Ёя дкZn2+,Cd2+ЕФШмвКжаЗжБ№МгШыNH3ЁЄH2OЃЌОљЩњГЩЧтбѕЛЏЮяГСЕэЃЌЕБNH3ЁЄH2OЙ§СПКѓЩњГЩАБЕФХфЮяЃК
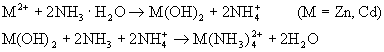
дкЫЎШмвКжаЃЌZn2+КЭCd2+гыЭЌжжХфЬхаЮГЩЕФСНжжХфКЯЮяЯрБШЃЌвЛАуЫЕРДКѓепНЯЮШЖЈЁЃ
Ёя Zn2+ЕФМјЖЈ
дкМюадЬѕМўЯТЃЌZn2+гыЖўБНСђыъЗДгІЃЌЩњГЩЗлКьЩЋЕФФкХфбЮГСЕэЃК
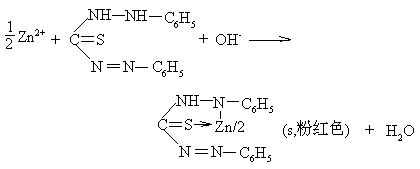
ДЫФкХфбЮФмШмгкCCl4жаЃЌГЪзиЩЋЁЃЪЕбщЯжЯѓЮЊЃКТЬЩЋЕФЖўБНСђыъЫФТШЛЏЬМШмвКгыZn2+ЗДгІКѓГфЗжеёЕДЃЌОВжУЃЌЩЯВуЮЊЗлКьЩЋЃЌЯТВуЮЊзиЩЋЁЃ
ЂВHg(Ђё),Hg(Ђђ)РызгЕФЗДгІ
Ёя ЫЎНтЗДгІ
дкHg(NO3)2КЭHg2(NO3)2ЕФЫсадШмвКжаЃЌЗжБ№гаЮоЩЋЕФ[Hg(H2O)6]2+КЭ[Hg2(H2O)x]2+ДцдкЃЌЫќУЧАДЯТЪНЫЎНтЃК
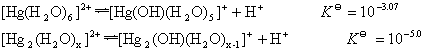
діДѓШмвКЕФЫсадПЩвжжЦЫќУЧЕФЫЎНтЁЃ
Ёя ГСЕэЗДгІЁЃдкHg2+КЭ ![]() ЕФШмвКжаМгШыЧПМюЪБЃЌЗжБ№ЩњГЩЛЦЩЋHgOКЭзиЩЋHg2OГСЕэ
ЕФШмвКжаМгШыЧПМюЪБЃЌЗжБ№ЩњГЩЛЦЩЋHgOКЭзиЩЋHg2OГСЕэ
Hg2OКмПьЗжНтЮЊHgOКЭHgЁЃ
Hg2O Ёњ HgO + Hg
ЫМПМЃКЕНФПЧАЮЊжЙЃЌЮвУЧбЇЙ§ФФаЉбєРызггыМюзїгУКѓЩњГЩбѕЛЏЮяЖјВЛЩњГЩЧтбѕЛЏЮяЁЃЮЊЪВУДЃП
Ёя дкHg2+ЃЌ ![]() ЕФШмвКжаЗжБ№МгШыЪЪСПЕФBr-,I-,
ЕФШмвКжаЗжБ№МгШыЪЪСПЕФBr-,I-, ![]() ,CN-КЭS2-ЪБЃЌЗжБ№ЩњГЩФбШмгкЫЎЕФЙЏбЮКЭбЧЙЏбЮЁЃЕЋаэЖрФбШмгкЫЎЕФбЧЙЏбЮМћЙтЪмШШШнвзЦчЛЏЮЊЯргІЕФЙЏбЮКЭЕЅжЪЙЏ(Г§Hg2Cl2)ЁЃР§ШчЃКЙЏбЮПЩШмгкЙ§СПЕФбєРызгШмвКаЮГЩХфКЯЮяЁЃ
,CN-КЭS2-ЪБЃЌЗжБ№ЩњГЩФбШмгкЫЎЕФЙЏбЮКЭбЧЙЏбЮЁЃЕЋаэЖрФбШмгкЫЎЕФбЧЙЏбЮМћЙтЪмШШШнвзЦчЛЏЮЊЯргІЕФЙЏбЮКЭЕЅжЪЙЏ(Г§Hg2Cl2)ЁЃР§ШчЃКЙЏбЮПЩШмгкЙ§СПЕФбєРызгШмвКаЮГЩХфКЯЮяЁЃ

HgI2ПЩШмгкЙ§СПЕФKIШмвКжаЃК
![]()
дкHgCl2ШмвКжаЭЈШыH2SЪБЃЌЛсЮіГіHgSГСЕэЃКР§ШчЃКЙЏбЮПЩШмгкЙ§СПЕФбєРызгШмвКаЮГЩХфКЯЮяЁЃ
![]()
HgSФбШмгкбЮЫсЛђЯѕЫсжаЃЌЕЋФмШмгкЙ§СПЕФХЈЕФNa2SШмвКжаЃК
![]()
ЪЕбщЪвГЃгУЭѕЫЎРДШмНтHgSЃК
![]()
Ёя Hg(Ђё),Hg(Ђђ)гыNH3ЁЄH2OЗДгІЯШЩњГЩNH2HgClАзЩЋГСЕэЃЌЕБгаЙ§СПЕФNH4ClДцдкЪБВХгыNH3аЮГЩХфКЯЮяЃК
HgCl2 + 2NH3 Ёњ NH2HgCl(s) + NH4Cl
Hg2Cl2 + 2NH3 Ёњ NH2Hg2Cl(s) + NH4Cl
NH2Hg2Cl Ёњ NH2HgCl + Hg
NH2HgCl + 2NH3 + NH4Cl Ёњ [Hg(NH3)4]Cl2
Ёя Hg2+ЕФМјЖЈЁЃ
РћгУSn2+ЕФЛЙдадЃЌМјЖЈHg2+ЕФДцдкЁЃ
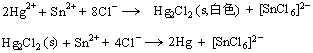
ШмвКгЩАзЁњКкЛвЁњКкЩЋЁЃРћгУИУдРэЃЌвВПЩгУHgCl2МјЖЈSn2+ЁЃ
ЫМПМЃКРћгУЭМЪОЗЈзмНсаПЁЂягЁЂЙЏЕФживЊаджЪ